2022
05-03
05-03
用超经典的mzmine2,对代谢物进行对峰
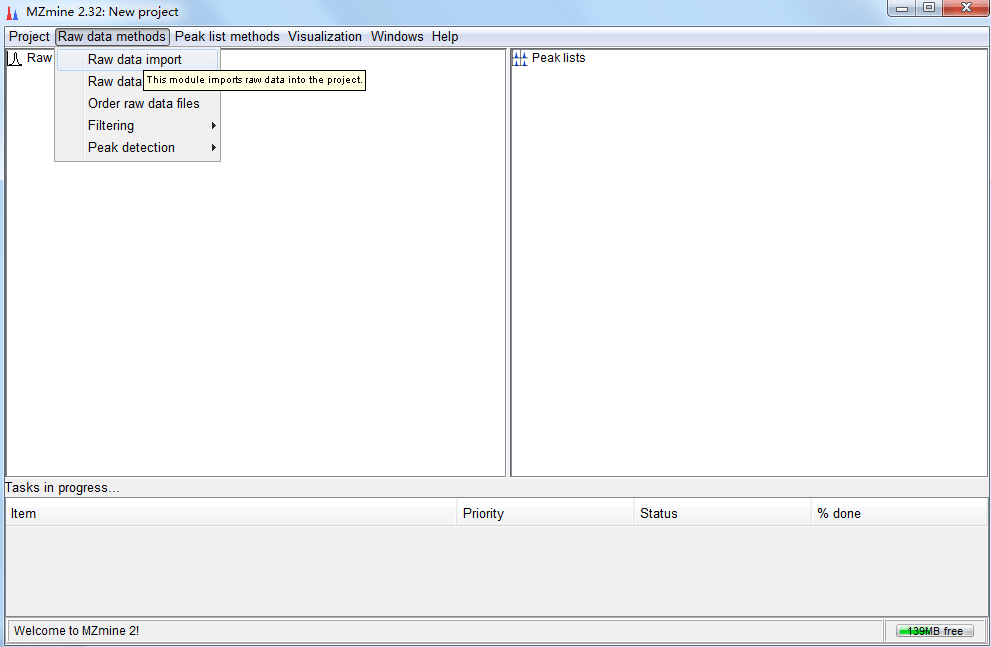 提到代谢组学峰比对,不得不提到一款老牌的比对软件,它就是MZmine2。它发表于2010年,前身是MZmine,发表于2006年,可谓是十分经典的一款代谢组学峰比对软件。MZmine 2的功能有很多,它是一款基于JAVA开发的开源软件,可以用于处理代谢组学profile数据,并且具有可视化和很多的分析功能。MZmine 2的一部分功能基于R,所以首先需要安装R环境,并且安装一些依赖包:instal... 阅 读 全 部 >
提到代谢组学峰比对,不得不提到一款老牌的比对软件,它就是MZmine2。它发表于2010年,前身是MZmine,发表于2006年,可谓是十分经典的一款代谢组学峰比对软件。MZmine 2的功能有很多,它是一款基于JAVA开发的开源软件,可以用于处理代谢组学profile数据,并且具有可视化和很多的分析功能。MZmine 2的一部分功能基于R,所以首先需要安装R环境,并且安装一些依赖包:instal... 阅 读 全 部 >
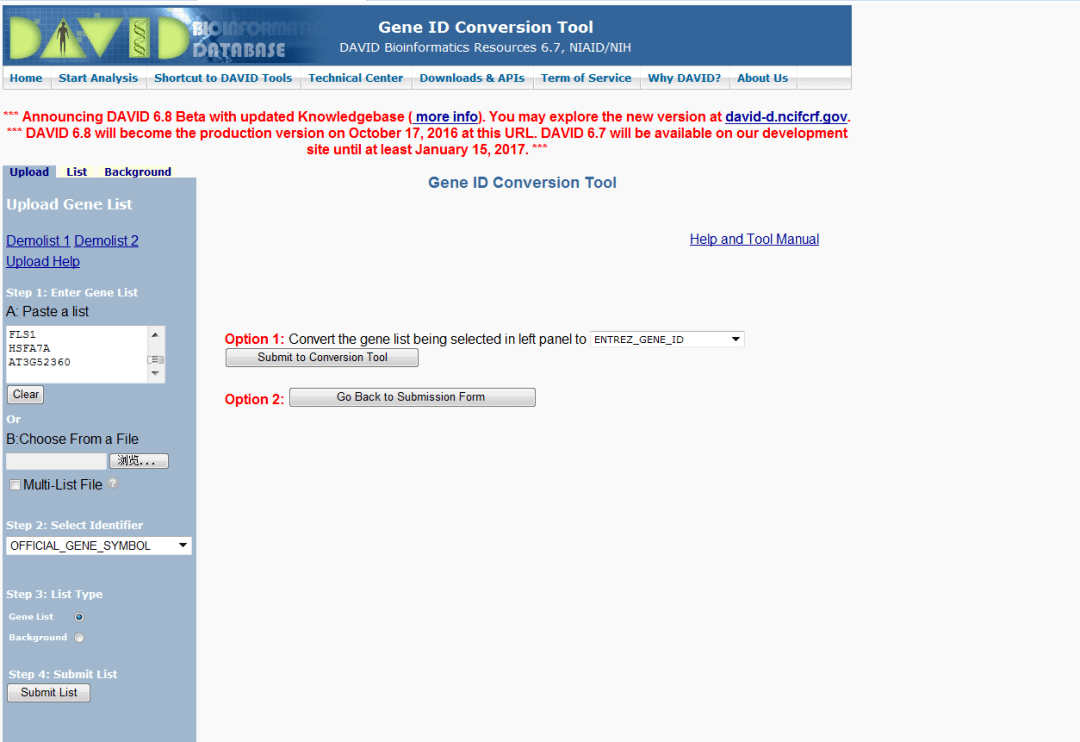 在进行差异基因表达分析时,得到显著差异基因后,接下来就需要分析这些基因参与了哪些功能,常见的就是GO功能注释和KEGG通路富集分析,今天就来介绍一下如何使用在线分析工具DAVID与KOBAS的进行KEGG通路富集分析。操作步骤进入DAVIDhttps://david.ncifcrf.gov/conversion.jsp上传基因列表选择物种开始分析选择转换选择格式,按照ENTREZ_GENE_ID转...
在进行差异基因表达分析时,得到显著差异基因后,接下来就需要分析这些基因参与了哪些功能,常见的就是GO功能注释和KEGG通路富集分析,今天就来介绍一下如何使用在线分析工具DAVID与KOBAS的进行KEGG通路富集分析。操作步骤进入DAVIDhttps://david.ncifcrf.gov/conversion.jsp上传基因列表选择物种开始分析选择转换选择格式,按照ENTREZ_GENE_ID转...  一般做完RNA的差异表达分析后,会有一堆的结果与数据。那么,怎么样让你的分析结果在paper中以更加直观形象(好看炫酷)的方式展示出来呢?前面我们已经出过【画图专题】史上最详细画热图函数pheatmap()参数讲解。下面小编介绍另外一种展示方式:火山图(Volcano Plot)。首先,我们要注意两个重要概念:FDR(False Discovery Rate)在差异表达分析过程中,采用统计学方法对...
一般做完RNA的差异表达分析后,会有一堆的结果与数据。那么,怎么样让你的分析结果在paper中以更加直观形象(好看炫酷)的方式展示出来呢?前面我们已经出过【画图专题】史上最详细画热图函数pheatmap()参数讲解。下面小编介绍另外一种展示方式:火山图(Volcano Plot)。首先,我们要注意两个重要概念:FDR(False Discovery Rate)在差异表达分析过程中,采用统计学方法对...  这个春天注定让人印象深刻,新型冠状病毒的爆发和蔓延牵动着亿万同胞的心,虽然疫情防控严峻复杂,但全国人民万众一心,终于取得了一定的阶段性的成果。今天小欧就为即将复工战斗在科研一线的小伙伴们送上福利,介绍一篇由加州大学圣地亚哥分校儿科和微生物创新中心、加州大学计算机科学与工程系和质谱创新中心合作,于2019年11月发表在Nature methods 上题为Learning represent...阅读...
这个春天注定让人印象深刻,新型冠状病毒的爆发和蔓延牵动着亿万同胞的心,虽然疫情防控严峻复杂,但全国人民万众一心,终于取得了一定的阶段性的成果。今天小欧就为即将复工战斗在科研一线的小伙伴们送上福利,介绍一篇由加州大学圣地亚哥分校儿科和微生物创新中心、加州大学计算机科学与工程系和质谱创新中心合作,于2019年11月发表在Nature methods 上题为Learning represent...阅读...  随着生物科技的迅速发展,每天都会有海量的生物学数据产生,如何有效的分析这些“生物学大数据”?生物信息学的应用变得尤为重要,在生物领域从基因测序,到基因编辑,再到基因疗法的精准医疗,由生物科技引发的又一场变革正悄然而至。试问大家做好准备迎接它到来了吗?本次分享的主题为:如何快速获取海量数据?我们就从物种的DNA或蛋白质序列说起,在我们的科学研究中下载序列是一件简单不过的事情,无非就是...阅读全文&...
随着生物科技的迅速发展,每天都会有海量的生物学数据产生,如何有效的分析这些“生物学大数据”?生物信息学的应用变得尤为重要,在生物领域从基因测序,到基因编辑,再到基因疗法的精准医疗,由生物科技引发的又一场变革正悄然而至。试问大家做好准备迎接它到来了吗?本次分享的主题为:如何快速获取海量数据?我们就从物种的DNA或蛋白质序列说起,在我们的科学研究中下载序列是一件简单不过的事情,无非就是...阅读全文&...  文章信息文章:An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics期刊:Nature Communications组学技术:scRNA-seq、mass spectrometry-based proteomics、bulk RNA-seq材料:scRNA-s...阅读全...
文章信息文章:An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics期刊:Nature Communications组学技术:scRNA-seq、mass spectrometry-based proteomics、bulk RNA-seq材料:scRNA-s...阅读全... 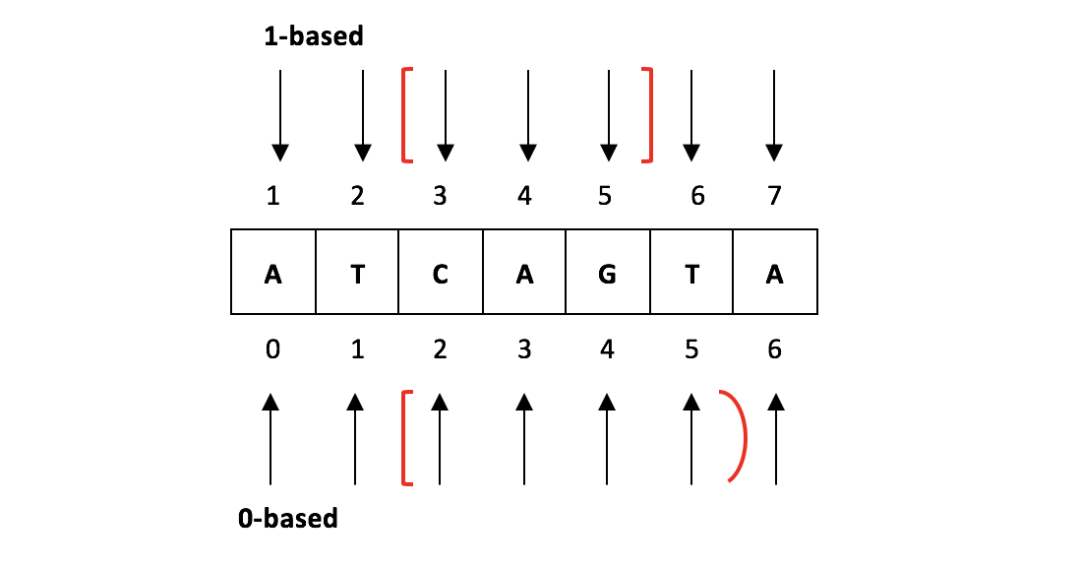 生信分析过程中,会与很多不同格式的文件打交道,除了原始测序数据 fastq 之外,还需要准备基因组文件 fasta 格式和基因注释文件 gtf 格式。在分析的过程中还会有众多中间文件的生成,如 bed 、 bed12 、 sam 、 bam 、 wig 、 bigwig 、 bedgraph 等,生成后我们一般会查看下内容了解文件每一列的含义,以此来决定需要提取哪些有用信息列来进行下...阅读全文...
生信分析过程中,会与很多不同格式的文件打交道,除了原始测序数据 fastq 之外,还需要准备基因组文件 fasta 格式和基因注释文件 gtf 格式。在分析的过程中还会有众多中间文件的生成,如 bed 、 bed12 、 sam 、 bam 、 wig 、 bigwig 、 bedgraph 等,生成后我们一般会查看下内容了解文件每一列的含义,以此来决定需要提取哪些有用信息列来进行下...阅读全文...  宏基因组(Metagenome)和宏转录组(Metatranscriptome)是通过鸟枪法测序技术(Shotgun sequencing),结合全微生物组关联分析(Microbiome-Wide Association Studies,MWAS)的策略,分别从DNA/RNA水平,全面精细地展示整个微生物群落的物种组成谱、功能代谢谱、表达谱,进而从原理上阐明微生物群落在生态系统中发挥...阅读全文...
宏基因组(Metagenome)和宏转录组(Metatranscriptome)是通过鸟枪法测序技术(Shotgun sequencing),结合全微生物组关联分析(Microbiome-Wide Association Studies,MWAS)的策略,分别从DNA/RNA水平,全面精细地展示整个微生物群落的物种组成谱、功能代谢谱、表达谱,进而从原理上阐明微生物群落在生态系统中发挥...阅读全文... 