2022
06-23
06-23
爱它就挖掘出它的最后价值:一个R包的全方位使用手册
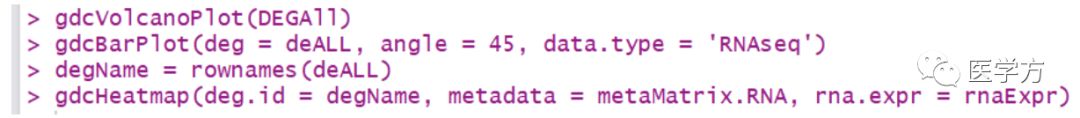 之前介绍了如何通过使用GDCRNATools这个包构建ceRNA网络,可以说整个过程非常简单和明了,对于我们学医的小朋友来讲,是个极好用的工具。但是这个包除了构建网络以外,还有很多附加的功能。要知道一篇ceRNA的文章,不单单只有那张网络图而已,还需要其他内容进行修饰和填充。那我们来看看这个包到底还有哪些功能?前两期链接:一这三条代码产生了下面几张图,具体就不解释了,一看就知道哈二输入上述的代码,... 阅 读 全 部 >
之前介绍了如何通过使用GDCRNATools这个包构建ceRNA网络,可以说整个过程非常简单和明了,对于我们学医的小朋友来讲,是个极好用的工具。但是这个包除了构建网络以外,还有很多附加的功能。要知道一篇ceRNA的文章,不单单只有那张网络图而已,还需要其他内容进行修饰和填充。那我们来看看这个包到底还有哪些功能?前两期链接:一这三条代码产生了下面几张图,具体就不解释了,一看就知道哈二输入上述的代码,... 阅 读 全 部 >
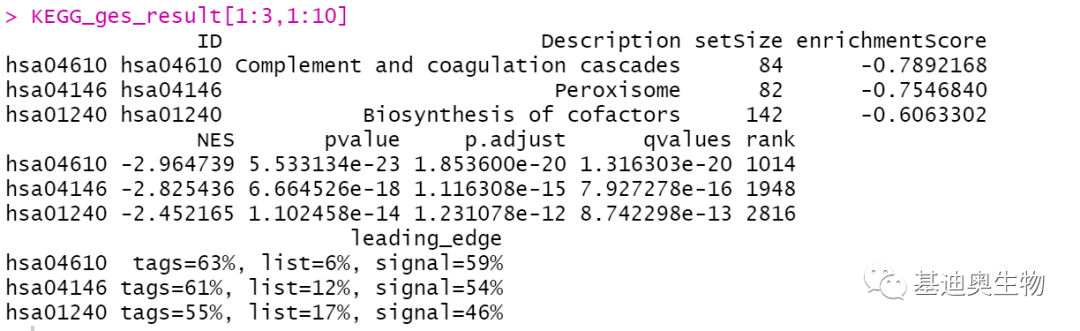 通过KEGG、GO等经典富集分析,我们能够了解到阈值筛选出的差异基因参与的通路和具备的功能,以及哪些功能/通路对表型变化是可能起主导作用的。但在经典富集分析中,我们无法得知某条通路下差异基因的总体变化趋势,即富集到同一通路下的基因既又上调也有下调,那么这条通路的表现形式到底是被激活了呢?还是被抑制了呢?且设定的阈值可能也会卡掉一些在统计学意义上无显著差异但实则有着重要生物学意义的基因,导致重要通路...
通过KEGG、GO等经典富集分析,我们能够了解到阈值筛选出的差异基因参与的通路和具备的功能,以及哪些功能/通路对表型变化是可能起主导作用的。但在经典富集分析中,我们无法得知某条通路下差异基因的总体变化趋势,即富集到同一通路下的基因既又上调也有下调,那么这条通路的表现形式到底是被激活了呢?还是被抑制了呢?且设定的阈值可能也会卡掉一些在统计学意义上无显著差异但实则有着重要生物学意义的基因,导致重要通路...  寻找差异表达的基因并识别它们的功能,是我们进行RNA测序的最主要目的。很明显,这些差异的基因必然与功能改变密切相关,例如,比较患病个体与正常个体的组织表达谱,不难想到这些显著失调的基因参与了生物学过程、信号通路等,导致了疾病的发生。前面已经讲了如何使用DESeq2、edgeR基于转录组测序获得的基因表达值鉴定差异表达基因。那么,后续如何继续通过生信分析的方法,探索差异表达的基因发挥...阅读全文&...
寻找差异表达的基因并识别它们的功能,是我们进行RNA测序的最主要目的。很明显,这些差异的基因必然与功能改变密切相关,例如,比较患病个体与正常个体的组织表达谱,不难想到这些显著失调的基因参与了生物学过程、信号通路等,导致了疾病的发生。前面已经讲了如何使用DESeq2、edgeR基于转录组测序获得的基因表达值鉴定差异表达基因。那么,后续如何继续通过生信分析的方法,探索差异表达的基因发挥...阅读全文&...  前面已经讲述了R包用clusterProfiler做GO富集分析clusterProfiler的GO富集分析方法,本篇继续演示R包goseq的GO富集分析。相比clusterProfiler中的GO富集分析方法,goseq的特别之处在于,不再使用超几何分布(Hyper-geometric distribution)检验,而是使用了Wallenius non-central hype...阅读全文&...
前面已经讲述了R包用clusterProfiler做GO富集分析clusterProfiler的GO富集分析方法,本篇继续演示R包goseq的GO富集分析。相比clusterProfiler中的GO富集分析方法,goseq的特别之处在于,不再使用超几何分布(Hyper-geometric distribution)检验,而是使用了Wallenius non-central hype...阅读全文&... 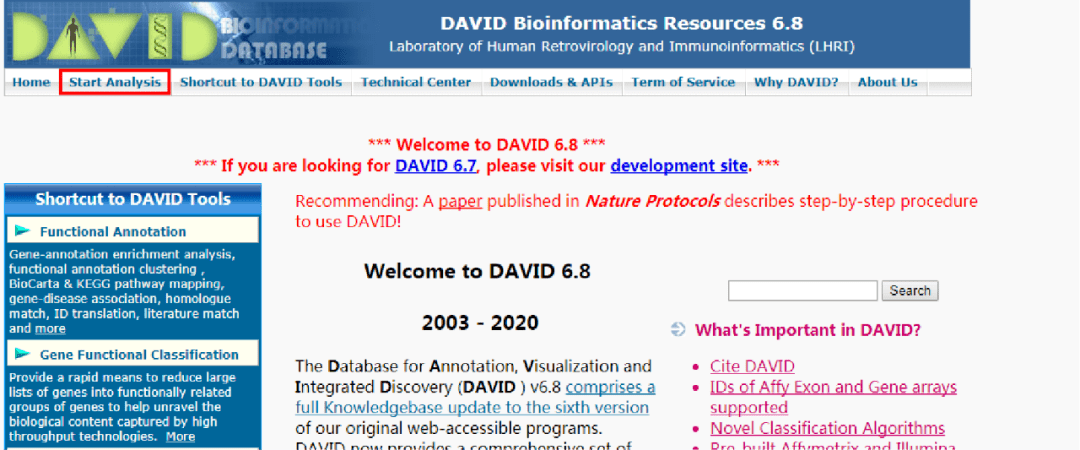 这期番茄君为大家带来如何利用DAVID在线数据库做GO与KEGG分析。1.点击文末“阅读全文”,获取DAVID在线数据库网址链接。打开后如下(部分):2.点击上图中红色框里的Start Analysis。如下: 3.上传基因。Step1: Enter Gene List有两个选项可以上传基因:第一种方法是在A: Paste a list的白色框里输入基因名字;第二种方法是利用B:Choose .....
这期番茄君为大家带来如何利用DAVID在线数据库做GO与KEGG分析。1.点击文末“阅读全文”,获取DAVID在线数据库网址链接。打开后如下(部分):2.点击上图中红色框里的Start Analysis。如下: 3.上传基因。Step1: Enter Gene List有两个选项可以上传基因:第一种方法是在A: Paste a list的白色框里输入基因名字;第二种方法是利用B:Choose .....  选自TowardsDataScience作者:Vihar Kurama参与:刘晓坤、许迪R 语言是结合了 S 编程语言的计算环境,可用于实现对数据的编程;它有很强大的数值分析工具,对于处理线性代数、微分方程和随机学的问题非常有用。通过一系列内建函数和库,你可以用 R 语言学习数据可视化,特别是它还有很多图形前端。本文将简单介绍 R 语言的编程基础,带你逐步实现第一个可视化案例。代码地址...阅读全...
选自TowardsDataScience作者:Vihar Kurama参与:刘晓坤、许迪R 语言是结合了 S 编程语言的计算环境,可用于实现对数据的编程;它有很强大的数值分析工具,对于处理线性代数、微分方程和随机学的问题非常有用。通过一系列内建函数和库,你可以用 R 语言学习数据可视化,特别是它还有很多图形前端。本文将简单介绍 R 语言的编程基础,带你逐步实现第一个可视化案例。代码地址...阅读全... 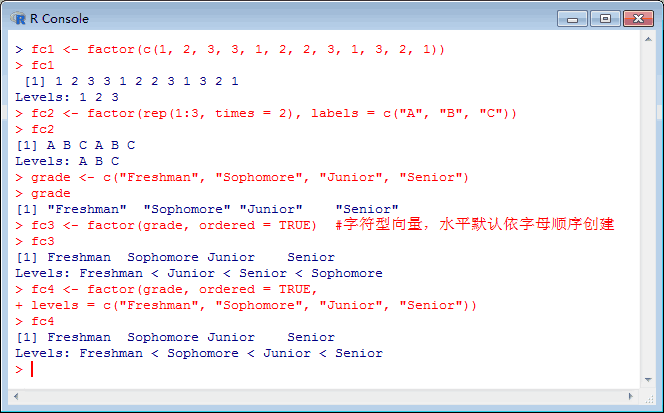 因子在R语言中可以用来表示名义型变量或有序变量。名义变量一般表示类别,如性别,种族等等。有序变量是有一定排序顺序的变量,如职称,年级等等。在R语言中,名义变量和有序变量可以使用因子来表示。创建因子在R语言中可以使用factor()函数和gl()函数来创建因子变量。(1)使用factor()函数factor()函数的语法格式为:f <- facto...阅读全文>>...
因子在R语言中可以用来表示名义型变量或有序变量。名义变量一般表示类别,如性别,种族等等。有序变量是有一定排序顺序的变量,如职称,年级等等。在R语言中,名义变量和有序变量可以使用因子来表示。创建因子在R语言中可以使用factor()函数和gl()函数来创建因子变量。(1)使用factor()函数factor()函数的语法格式为:f <- facto...阅读全文>>...  这些周R语言学习,记录如下。01数据操作概述数据操作是一个对数据加工处理以满足后续数据工作(分析或者建模)的过程。数据操作主要做的事情,包括:数据列的操作。数据行的操作。数据的聚合操作。数据的其它操作。我用dplyr包做数据操作,它功能强大,应用简便,编码易懂。dplyr包做各种数据操作,无论多复杂,都可以分解5种基...阅读全文>>...
这些周R语言学习,记录如下。01数据操作概述数据操作是一个对数据加工处理以满足后续数据工作(分析或者建模)的过程。数据操作主要做的事情,包括:数据列的操作。数据行的操作。数据的聚合操作。数据的其它操作。我用dplyr包做数据操作,它功能强大,应用简便,编码易懂。dplyr包做各种数据操作,无论多复杂,都可以分解5种基...阅读全文>>...  派森诺转录组致力于科技服务的产品丰富且专业,产品在丰富的同时也有细致划分。今天带大家来了解一下对于无参考基因组的物种,比较转录组和无参转录组的区别。区别:1、分析目的不同:无参转录组测序:利用高通量测序技术进行cDNA测序,全面快速地获取某一种特定器官或组织在某一状态下的几乎所有转录本。根据将不同组的unigene进行比较,得到差异基因进行分析。比较转录组测序:是基于高通...阅读全文>&g...
派森诺转录组致力于科技服务的产品丰富且专业,产品在丰富的同时也有细致划分。今天带大家来了解一下对于无参考基因组的物种,比较转录组和无参转录组的区别。区别:1、分析目的不同:无参转录组测序:利用高通量测序技术进行cDNA测序,全面快速地获取某一种特定器官或组织在某一状态下的几乎所有转录本。根据将不同组的unigene进行比较,得到差异基因进行分析。比较转录组测序:是基于高通...阅读全文>&g...  转载请注明:解螺旋·临床医生科研成长平台10余年前,华盛顿大学的生物学教授Michael Skinner的团队做过一系列有意思的实验。2005年,他们将怀孕的女大鼠(F0)暴露于一种农业中常用的杀真菌剂,乙烯菌核利。接着在无杀菌剂的环境中将她们的后代拉扯大,一直养了4代,也就是养到了她们的玄孙(F4)。结果几乎所有玄孙子的精子数量、繁殖能力都有下降,并且出现了年龄相关的不育症。在所...阅读全文&...
转载请注明:解螺旋·临床医生科研成长平台10余年前,华盛顿大学的生物学教授Michael Skinner的团队做过一系列有意思的实验。2005年,他们将怀孕的女大鼠(F0)暴露于一种农业中常用的杀真菌剂,乙烯菌核利。接着在无杀菌剂的环境中将她们的后代拉扯大,一直养了4代,也就是养到了她们的玄孙(F4)。结果几乎所有玄孙子的精子数量、繁殖能力都有下降,并且出现了年龄相关的不育症。在所...阅读全文&... 