2022
07-09
07-09
16S从实验到数据分析最全流程
 本文主要介绍了16S的实验、建库、数据分析等过程,也是我自己近期的一个小总结,初学之时从很多前辈的无私分享中受益良多,在此也和大家分享一些我的见解,当然我也只是一个初学者,还有很多不完备之处,希望能与各位一起交流分享。导航本文一共分为三个部分:实验部分建库测序16S测序数据分析一. 实验部分:DNA提取与质检1...阅读全文>>... 阅 读 全 部 >
本文主要介绍了16S的实验、建库、数据分析等过程,也是我自己近期的一个小总结,初学之时从很多前辈的无私分享中受益良多,在此也和大家分享一些我的见解,当然我也只是一个初学者,还有很多不完备之处,希望能与各位一起交流分享。导航本文一共分为三个部分:实验部分建库测序16S测序数据分析一. 实验部分:DNA提取与质检1...阅读全文>>... 阅 读 全 部 >
本文主要介绍了16S的实验、建库、数据分析等过程,也是我自己近期的一个小总结,初学之时从很多前辈的无私分享中受益良多,在此也和大家分享一些我的见解,当然我也只是一个初学者,还有很多不完备之处,希望能与各位一起交流分享。导航本文一共分为三个部分:实验部分建库测序16S测序数据分析一. 实验部分:DNA提取与质检1...阅读全文>>... 阅 读 全 部 >
本文主要介绍了16S的实验、建库、数据分析等过程,也是我自己近期的一个小总结,初学之时从很多前辈的无私分享中受益良多,在此也和大家分享一些我的见解,当然我也只是一个初学者,还有很多不完备之处,希望能与各位一起交流分享。导航本文一共分为三个部分:实验部分建库测序16S测序数据分析一. 实验部分:DNA提取与质检1...阅读全文>>... 阅 读 全 部 >
 参考链接:https://ccb.jhu.edu/software/stringtie/index.shtml?t=manual#input参数简介StringTie的基本用法: stringtie <aligned_reads.bam> [options]*其中,aligned_reads.bam 是输入文件,该输入文件要求必须按其基因组位置排序,如TopHat...阅读全文>... 阅 读 全 部 >
参考链接:https://ccb.jhu.edu/software/stringtie/index.shtml?t=manual#input参数简介StringTie的基本用法: stringtie <aligned_reads.bam> [options]*其中,aligned_reads.bam 是输入文件,该输入文件要求必须按其基因组位置排序,如TopHat...阅读全文>... 阅 读 全 部 >
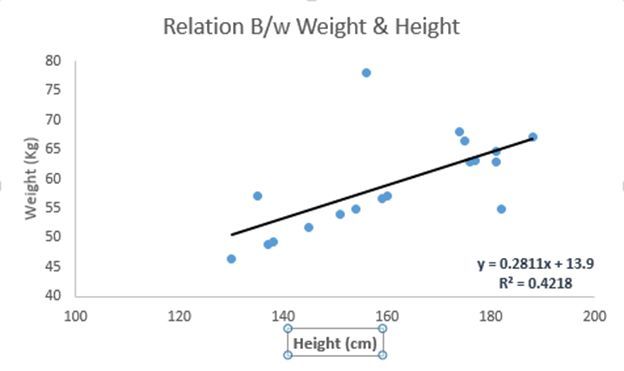 转自:TalkingData原文作者:Sunil Ray译者:TalkingData 张永超原文链接:https://www.analyticsvidhya.com/blog/2017/09/common-machine-learning-algorithms/“简介”从广义上讲,机器学习算法有三种类型:监督学习该算法是由一个目标/结果变量(也成为因变量)组成,该变量可以从一组给定的预测...阅读... 阅 读 全 部 >
转自:TalkingData原文作者:Sunil Ray译者:TalkingData 张永超原文链接:https://www.analyticsvidhya.com/blog/2017/09/common-machine-learning-algorithms/“简介”从广义上讲,机器学习算法有三种类型:监督学习该算法是由一个目标/结果变量(也成为因变量)组成,该变量可以从一组给定的预测...阅读... 阅 读 全 部 >
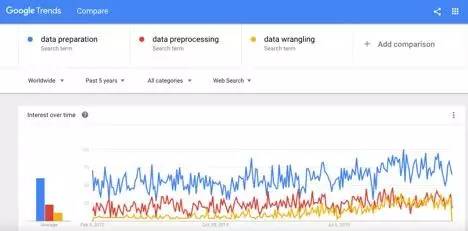 翻译|谢旭审校|张卫滨本文比较了用于数据准备的几种方法,它们分别是提取-变换-加载批处理(ETL)、流式获取和数据整理。本文还讨论了数据准备如何与可视化分析相关联,以及不同用户角色(如数据科学家或业务分析人员)应如何共同构建分析模型的最佳实践。要点在常见的机器学习/深度学习项目里,数据准备占去整个分析管道的60%到80%。市场上有...阅读全文>>... 阅 读 全 部 >
翻译|谢旭审校|张卫滨本文比较了用于数据准备的几种方法,它们分别是提取-变换-加载批处理(ETL)、流式获取和数据整理。本文还讨论了数据准备如何与可视化分析相关联,以及不同用户角色(如数据科学家或业务分析人员)应如何共同构建分析模型的最佳实践。要点在常见的机器学习/深度学习项目里,数据准备占去整个分析管道的60%到80%。市场上有...阅读全文>>... 阅 读 全 部 >
 grep 'Overall alignment rate: ' *.log |sed 's/Overall alignment rate: //'|sed 's/.log://'可以重定向阅读全文>>... 阅 读 全 部 >
grep 'Overall alignment rate: ' *.log |sed 's/Overall alignment rate: //'|sed 's/.log://'可以重定向阅读全文>>... 阅 读 全 部 >
 测试开头爸妈: 今年不回来么?1引言没错, 就是美国大名鼎鼎的航天局 NASA, 2021 年 4 月 23 日 在 iScience 期刊上发表了一篇处理 RNA-seq 数据的一篇文章。这篇文章提供了标准分析的一些代码,并且采用了 ENCODE 计划中的参考代码。这个 pipeline 主要包括了 quality control, read trimming,...阅读全文>>... 阅 读 全 部 >
测试开头爸妈: 今年不回来么?1引言没错, 就是美国大名鼎鼎的航天局 NASA, 2021 年 4 月 23 日 在 iScience 期刊上发表了一篇处理 RNA-seq 数据的一篇文章。这篇文章提供了标准分析的一些代码,并且采用了 ENCODE 计划中的参考代码。这个 pipeline 主要包括了 quality control, read trimming,...阅读全文>>... 阅 读 全 部 >
 RNA-seq 即转录组测序技术,就是用高通量测序技术进行测序分析,反映出 mRNA,smallRNA,noncodingRNA 等或者其中一些的表达水平,寻找表达差异的基因预测或验证相关的分子机制及功能。2016 年发表在 nature protocols 上一篇关于转录本精确定量[1]的文章:文章中以 HISAT + Stringtie + Ballgrown 的...阅读全文>>... 阅 读 全 部 >
RNA-seq 即转录组测序技术,就是用高通量测序技术进行测序分析,反映出 mRNA,smallRNA,noncodingRNA 等或者其中一些的表达水平,寻找表达差异的基因预测或验证相关的分子机制及功能。2016 年发表在 nature protocols 上一篇关于转录本精确定量[1]的文章:文章中以 HISAT + Stringtie + Ballgrown 的...阅读全文>>... 阅 读 全 部 >
 蛋白质互作网络是由蛋白通过彼此之间的相互作用构成,来参与生物信号传递、基因表达调节、能量和物质代谢及细胞周期调控等生命过程的各个环节。系统分析大量蛋白在生物系统中的相互作用关系,对了解生物系统中蛋白质的工作原理,了解疾病等特殊生理状态下生物信号和能量物质代谢的反应机制,以及了解蛋白之间的功能联系都有重要意义。那么如何分析蛋白质互作网络?如何发现复杂网络的关键蛋白和子网络?如何使用马尔科夫(MCL)... 阅 读 全 部 >
蛋白质互作网络是由蛋白通过彼此之间的相互作用构成,来参与生物信号传递、基因表达调节、能量和物质代谢及细胞周期调控等生命过程的各个环节。系统分析大量蛋白在生物系统中的相互作用关系,对了解生物系统中蛋白质的工作原理,了解疾病等特殊生理状态下生物信号和能量物质代谢的反应机制,以及了解蛋白之间的功能联系都有重要意义。那么如何分析蛋白质互作网络?如何发现复杂网络的关键蛋白和子网络?如何使用马尔科夫(MCL)... 阅 读 全 部 >
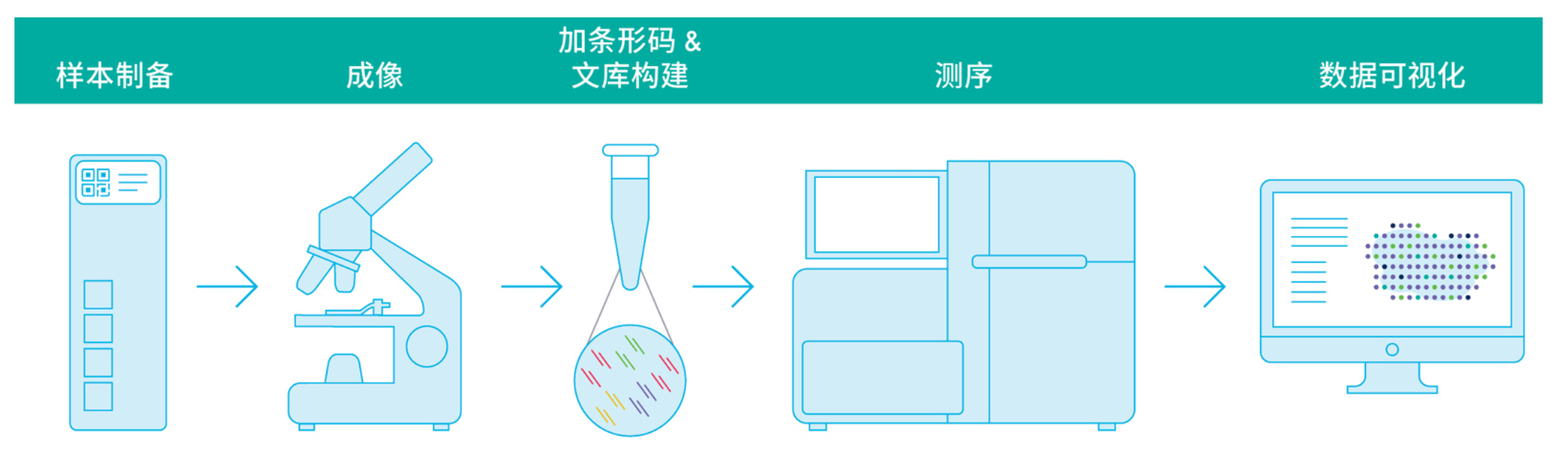 把病理切片裂解制备成单细胞悬液,得到了“是什么”,却丢失了“在哪里”。如何兼得类型和位置信息呢?follow me 跟小编一起解开空间转录组技术的面纱吧。在生命科学研究的道路上,历经了多组学联合覆盖“面“的,单细胞技术涉及“点“的研究历程,为了更加深入的对生命机制,疾病机理等研究,科学家不断发明新的技术,丰富新的维度,空间转录组技术也由此得到了很大的提升,操作上由繁至简,技术上由少...阅读全文&... 阅 读 全 部 >
把病理切片裂解制备成单细胞悬液,得到了“是什么”,却丢失了“在哪里”。如何兼得类型和位置信息呢?follow me 跟小编一起解开空间转录组技术的面纱吧。在生命科学研究的道路上,历经了多组学联合覆盖“面“的,单细胞技术涉及“点“的研究历程,为了更加深入的对生命机制,疾病机理等研究,科学家不断发明新的技术,丰富新的维度,空间转录组技术也由此得到了很大的提升,操作上由繁至简,技术上由少...阅读全文&... 阅 读 全 部 >

生信圈
欢迎您支持我的公众号
点击此处可关闭