2021
10-05
10-05
R Markdown:可能是你数据分析报告最好的解决方案
 为什么要用Rmd?“如果你是第一次听到R Markdown这个名词的话,可能你会问R Markdown是什么?我们能用它来做什么?怎么使用R Markdown?且听小编跟你道来。”当初人才计划进行到第二阶段的时候,水妈要求我们所有的 R 代码都需要通过R Markdown生成html来提交。一开始并不了解其中奥义,在此之前印象中那只是一个可以用来生成数据分析文件格式的工具。经过一段时间学...阅读... 阅 读 全 部 >
为什么要用Rmd?“如果你是第一次听到R Markdown这个名词的话,可能你会问R Markdown是什么?我们能用它来做什么?怎么使用R Markdown?且听小编跟你道来。”当初人才计划进行到第二阶段的时候,水妈要求我们所有的 R 代码都需要通过R Markdown生成html来提交。一开始并不了解其中奥义,在此之前印象中那只是一个可以用来生成数据分析文件格式的工具。经过一段时间学...阅读... 阅 读 全 部 >
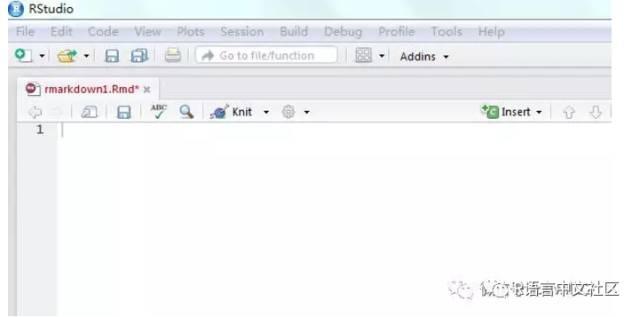 R Markadown 作为一款通过R语言创建动态文档的写作排版工具,为数据科学提供了现成的写作框架。通过 R Markdown 不仅可以运行和保存R代码,还可以生成高质量的数据分析报告并以HTML、PDF或者word的形式分享。1,get started很早就对R语言可以制作高质量的报告有所耳闻,但也没有很强的意愿去研究一番。究其缘由在于写公众号时无法不会优雅的插入代码块,便有意...阅读全文&...
R Markadown 作为一款通过R语言创建动态文档的写作排版工具,为数据科学提供了现成的写作框架。通过 R Markdown 不仅可以运行和保存R代码,还可以生成高质量的数据分析报告并以HTML、PDF或者word的形式分享。1,get started很早就对R语言可以制作高质量的报告有所耳闻,但也没有很强的意愿去研究一番。究其缘由在于写公众号时无法不会优雅的插入代码块,便有意...阅读全文&... 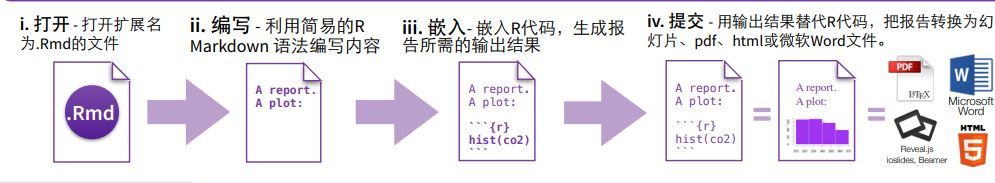 1 如何用Rmarkdown生成中文自动化报告?本文总结Rmarkdown的使用,主要回答以下问题:Rmarkdown是什么?如何使用Rmarkdown?如何使用Rmarkdown生成中文文档?Rmarkdown应用举例?Rmarkdown是什么?Analyze. Share. Reproduce.Your data tel...阅读全文>>...
1 如何用Rmarkdown生成中文自动化报告?本文总结Rmarkdown的使用,主要回答以下问题:Rmarkdown是什么?如何使用Rmarkdown?如何使用Rmarkdown生成中文文档?Rmarkdown应用举例?Rmarkdown是什么?Analyze. Share. Reproduce.Your data tel...阅读全文>>... 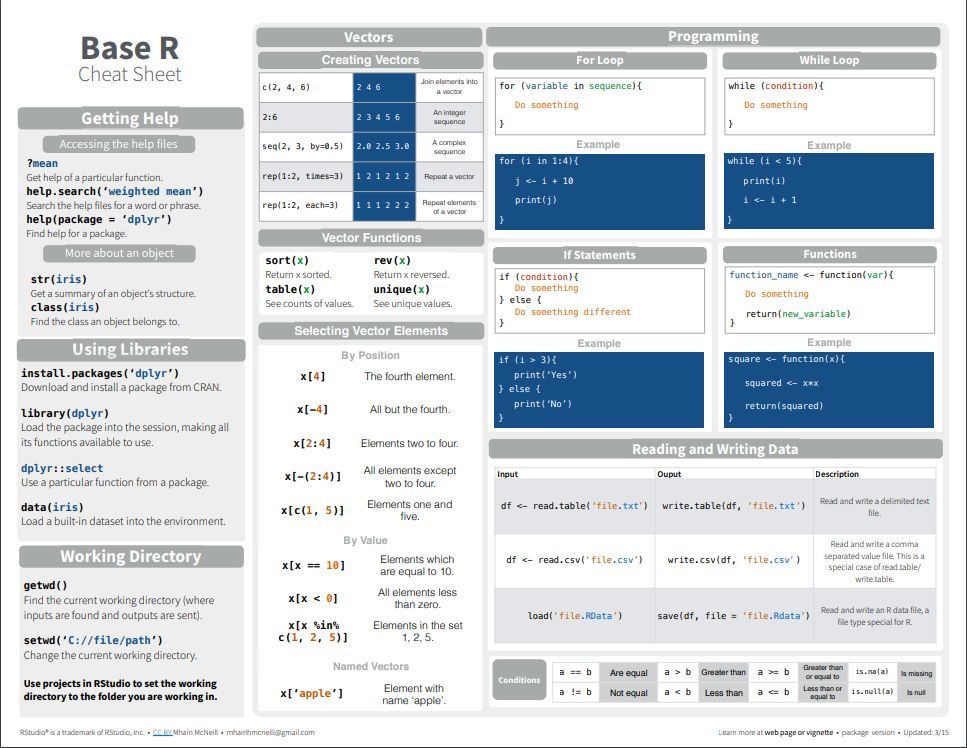 编者按:R知识速查表囊括R基本知识,R高级知识,R数据导入,R数据可视化,R数据处理,字符串处理,正则表达式,日期时间处理,数据转换和机器学习。对于R新手,建议按着这个顺序学习和实践。【温馨提示:点击图片,可查看大图】一:基本 R 知识二:高级R知识三:数据可视化知识四:数据导入知识五:数据处理知识六:数据转换知识七:字符串处理知识八:正则表达式知识九:日期时间处理十:机器学习知识参考资料:......
编者按:R知识速查表囊括R基本知识,R高级知识,R数据导入,R数据可视化,R数据处理,字符串处理,正则表达式,日期时间处理,数据转换和机器学习。对于R新手,建议按着这个顺序学习和实践。【温馨提示:点击图片,可查看大图】一:基本 R 知识二:高级R知识三:数据可视化知识四:数据导入知识五:数据处理知识六:数据转换知识七:字符串处理知识八:正则表达式知识九:日期时间处理十:机器学习知识参考资料:......  如果你对生物信息不那么了解,或许会搞混FASTQ和FASTA,它们都是序列保存的一种格式,是用于NGS分析的基础数据。其实二者很好区分,并且可以通过一定的处理进行格式转换。FASTQFASTQ:是基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式。你拿到的测序数据均是fastq格式的。其序列以及质量信息都是使用一个ASCII字符标示,最初是由Sanger开发,目的是将F...阅读...
如果你对生物信息不那么了解,或许会搞混FASTQ和FASTA,它们都是序列保存的一种格式,是用于NGS分析的基础数据。其实二者很好区分,并且可以通过一定的处理进行格式转换。FASTQFASTQ:是基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式。你拿到的测序数据均是fastq格式的。其序列以及质量信息都是使用一个ASCII字符标示,最初是由Sanger开发,目的是将F...阅读...  在实际工作中,每个数据科学项目各不相同,但基本都遵循一定的通用流程。具体如下:数据科学工作流程数据导入数据整理反复理解数据数据可视化数据转换统计建模作出推断(比如预测)沟通交流自动化分析程序开发下面列出每个步骤最有用的一些R包:数据导入以下R包主要用于数据导入和保存数据feather:一种快速,轻量级的文件格式。在R和python上都可使用readr:实现表格数据的快...阅读全文>>...
在实际工作中,每个数据科学项目各不相同,但基本都遵循一定的通用流程。具体如下:数据科学工作流程数据导入数据整理反复理解数据数据可视化数据转换统计建模作出推断(比如预测)沟通交流自动化分析程序开发下面列出每个步骤最有用的一些R包:数据导入以下R包主要用于数据导入和保存数据feather:一种快速,轻量级的文件格式。在R和python上都可使用readr:实现表格数据的快...阅读全文>>... 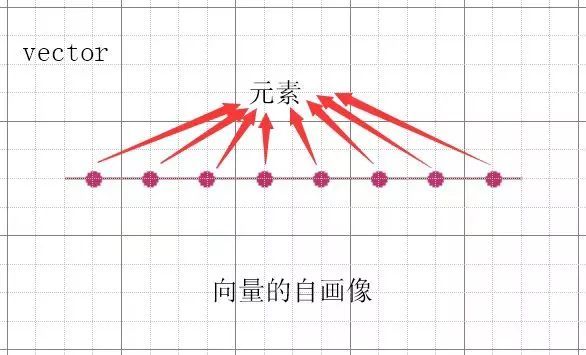 前文我们讲到R处理数据面对的6种对象:向量,矩阵,数组,因子,列表,数据框。A. 那我们就得好好给大家介绍一下这位能者的6个对象都长什么样子了。· 1.向量 ·向量大体上分为3种,数值向量,字符向量,逻辑向量。(单个向量内元素类型必须一致)数值向量:> c(1,2,3,4,5,6,7)[1] 1 2 3 4 5 6 7字符向量(字符向量使用单...阅读全文>>...
前文我们讲到R处理数据面对的6种对象:向量,矩阵,数组,因子,列表,数据框。A. 那我们就得好好给大家介绍一下这位能者的6个对象都长什么样子了。· 1.向量 ·向量大体上分为3种,数值向量,字符向量,逻辑向量。(单个向量内元素类型必须一致)数值向量:> c(1,2,3,4,5,6,7)[1] 1 2 3 4 5 6 7字符向量(字符向量使用单...阅读全文>>... 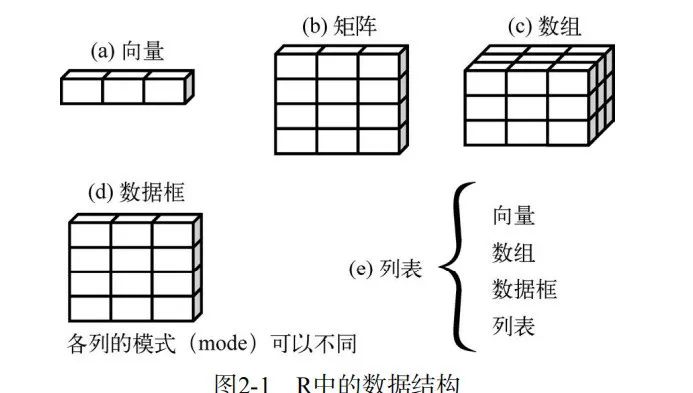 预计阅读时间4分钟R拥有许多用于存储数据的对象类型,包括标量、向量、矩阵、数组、数据框和列表。它们在存储数据的类型、创建方式、结构复杂度,以及用于定位和访问其中个别元素的标记等方面均有所不同。下图是一张R语言数据结构图。文章目录快速检索,先看看哪部分感兴趣,查漏补缺~向量向量类型向量的创建向量中元素的访...阅读全文>>...
预计阅读时间4分钟R拥有许多用于存储数据的对象类型,包括标量、向量、矩阵、数组、数据框和列表。它们在存储数据的类型、创建方式、结构复杂度,以及用于定位和访问其中个别元素的标记等方面均有所不同。下图是一张R语言数据结构图。文章目录快速检索,先看看哪部分感兴趣,查漏补缺~向量向量类型向量的创建向量中元素的访...阅读全文>>...  1.环境:CentOS7+已安装好miniconda 2.conda安装wget<pre>conda install wget</pre>3.下载conda环境文件<pre>wget https://data.qiime2.org/distro/core...阅读全文>>...
1.环境:CentOS7+已安装好miniconda 2.conda安装wget<pre>conda install wget</pre>3.下载conda环境文件<pre>wget https://data.qiime2.org/distro/core...阅读全文>>...  微生物宏基因组是当今世界最热门的科研领域之一。越来越多的研究表明,人体微生物发挥着重要的健康作用,但大部分多样性仍未得到充分探索,尤其是在除肠道以外的身体部位及非西方人群。在今日发表在Cell期刊的一项研究中,由意大利特伦托大学NicolaSegata领导的研究团队利用来自不同地理位置、生活方式和年龄人群的9,428个宏基因组,突破性地重建了154,723个人体微生物基因组(45%高质量),其.....
微生物宏基因组是当今世界最热门的科研领域之一。越来越多的研究表明,人体微生物发挥着重要的健康作用,但大部分多样性仍未得到充分探索,尤其是在除肠道以外的身体部位及非西方人群。在今日发表在Cell期刊的一项研究中,由意大利特伦托大学NicolaSegata领导的研究团队利用来自不同地理位置、生活方式和年龄人群的9,428个宏基因组,突破性地重建了154,723个人体微生物基因组(45%高质量),其..... 