利用16S/ITS多样性测序,可以准确了解群落的微生物组成,同时也可以大概的了解群落功能组成情况。因为16S、ITS的功能预测能在一定程度上代替宏基因组、宏转录组的群落功能的分析,所以是现在微生物比较热门的分析点。
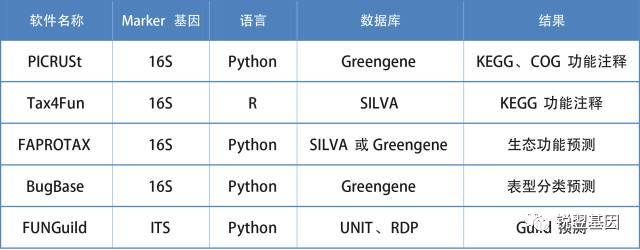
16S的功能预测有四个相关软件,分别是PICRUSt[1]、Tax4Fun[2]、FAPROTAX[3]、BugBase[4]
ITS的功能预测相关软件是FUNGuild[5]
这些功能预测软件都有自己的特色和侧重点。通过下边的表格可以大概了解各个软件之间的区别:
1. PICRUSt
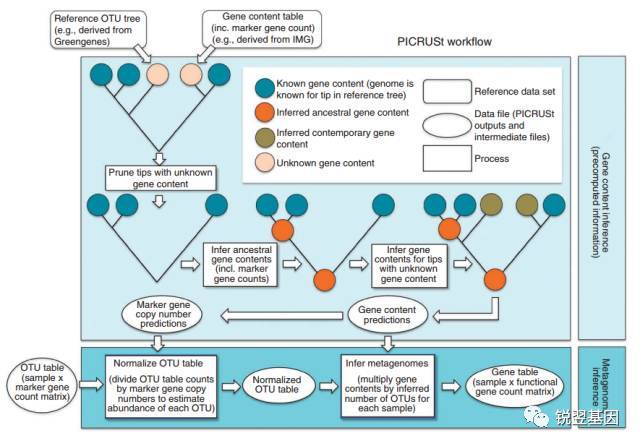
PICRUSt的全称是Phylogenetic Investigation of Communities by Reconstruction of Unobserved States。它基于Greengene数据库中OTU的tree,和OTU上的基因信息,推断它们的共同祖先的基因功能谱,同时对Greengenes数据库中其它未测物种的基因功能谱进行推断,构建古菌和细菌域全谱系的基因功能预测谱,最后将测序得到的菌群组成“映射”到数据库中,就能进行菌群代谢功能预测了。
软件原理图
安装PICRUSt
在linux下安装PICRUSt软件非常简单。
请参考官方文档 https://picrust.github.io/picrust/install.html#install
PICRUSt软件使用分4步
> Step 1:拥有一个完整OTU丰度表, 如实例中的tutorials/hmp_mock_16S.biom.
> Step 2:标准化 OTU Table(OTU丰度表)
normalize_by_copy_number.py脚本根据已知微生物的16s拷贝数,进行标准化OTU丰度表。
输入:OTU的丰度表(基于Greengenes数据库)
输出:标准化之后的OTU丰度表
normalize_by_copy_number.py -i your_otu_table.biom
-o normalized_otus.biom
> Step 3: 菌群的功能预测
predict_metagenomes.py 脚本分析菌群的功能预测。它把标准化OTU注释上的所有功能特征都相加,得到一个行是功能、列是样品的丰度矩阵表。
输入:通过normalize_by_copy_number.py脚本得到的标准化OTU丰度表
输出:功能丰度表
predict_metagenomes.py
-i normalized_otus.biom
-o metagenome_predictions.biom
如果使用了-f参数,就可以输出制表符割开的丰度表
predict_metagenomes.py
-f
-i normalized_otus.biom
-o metagenome_predictions.tab
(Optional) NSTI values for each sample can be obtained using the -a option. (We strongly recommend this step, as NSTI values are precalculated for common inputs):
predict_metagenomes.py
-i normalized_otus.biom
-o metagenome_predictions.tab
-a nsti_per_sample.tab
参数--gg_version可以使用旧版本的Greengene数据库
predict_metagenomes.py
--gg_version 18may2012
-i normalized_otus.biom
-o metagenome_predictions.biom
参数--type_of_prediction有三个选项ko, cog, rfam ,默认是ko
predict_metagenomes.py
--type_of_prediction cog
-i normalized_otus.biom
-o metagenome_predictions.biom
> Step 4: 分析菌群功能预测的结果
现在你已经拥有了菌群功能预测结果了,你可以按照普通宏基因数据分析方法进行后续分析了。
例如categorize_by_function.py脚本进行pathway的分析;metagenome_contributions.py脚本分析具体哪些OTUs正在为特定的功能做出贡献;也可以是HUManN、LEfSe和GraPhlAn软件进行分析(点击软件名字,可查看相应教程)。
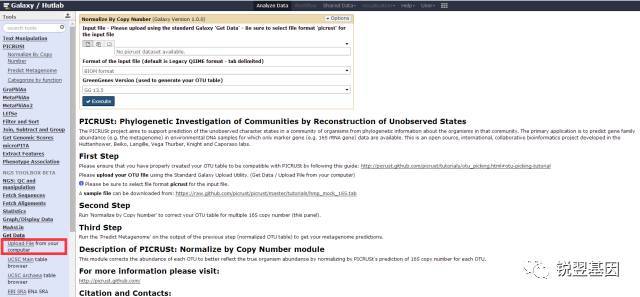
在线分析
PICRUSt的在线分析软件网址http://huttenhower.sph.harvard.edu/galaxy/root?tool_id=lefse_upload
首先,上传做功能预测的biom格式的文件,或是制表符分割的tsv文件。
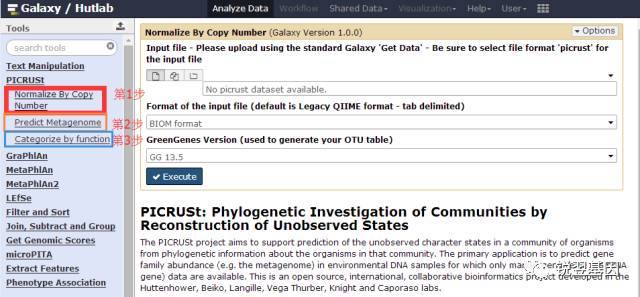
然后只要简单的依次运行1、2、3步就可以了。依次能得到标准化的OTU丰度表、对群落功能预测结果、功能的分类结果。
2. Tax4Fun
Tax4Fun是基于SILVA数据库,将16S的OTU信息进行功能预测的软件。它和PICRUSt软件比较类似。Tax4Fun的文献研究表明它在功能预测方面优于PICRUSt软件。下图是文献的原图:
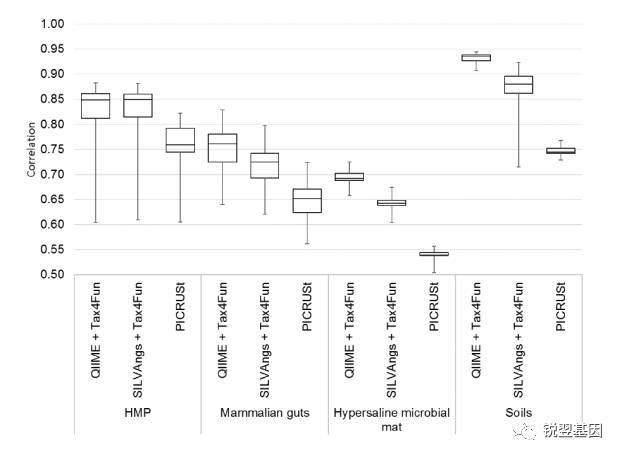
Tax4Fun的相关性的数值都高于PICRUSt结果,总体分布在0.64~0.87范围内。一定程度上也说明了,16S的功能预测和实际的功能是有区别的。
软件安装
Tax4Fun是一个R语言的扩展包,下载网址:http://tax4fun.gobics.de/
R CMD INSTALL Tax4Fun_0.3.1.tar.gz
软件使用
library("Tax4Fun")
file <- "GNM_0.97_table.txt"#官方的example数据
QIIMESingleData <- importQIIMEData(file)
folderReferenceData <- "~/Downloads/Tax4FunData/SILVA119/"#需要把下载SILVA数据库解压
Tax4FunOutput <- Tax4Fun(QIIMESingleData, folderReferenceData)
print(Tax4FunOutput$Tax4FunProfile)#这个输出的是功能丰度表
到这里我们已经能够得到kegg功能的丰度表,你可以按照普通宏基因数据分析方法进行后续分析了。
3. FAPROTAX
FAPROTAX全称是Functional Annotation of Prokaryotic Taxa。它是原核的微生物(例如属或者种)注释代谢或其他生态相关的功能(例如硝化,反硝化,发酵)的一个数据库和软件。FAPROTAX预测的功能主要集中在海洋、湖泊中微生物的功能,特别是硫、碳、氢、氮的循环功能。它有一个名为collapse_table.py的脚本,根据OTU的丰度表,计算出功能的丰度表。这是一个比较小范围的数据库,所以使用者不太多。
下面的原理图很清晰地展示了输入、输出的结果。
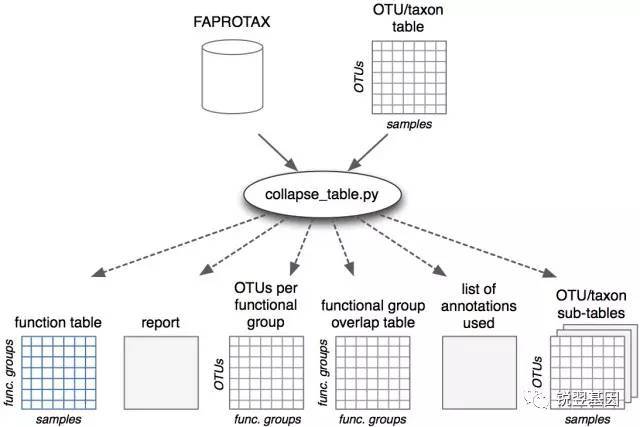
输入:OTU的丰度表
输出:样品功能丰度表,报告,OTU功能丰度表……
软件安装
数据库及脚本下载的地址:
http://www.zoology.ubc.ca/louca/FAPROTAX/lib/php/index.php?section=Download
软件使用
cd/home/shared/predict_pheno/FAPROTAX_1.0/example_input
collapse_table.py -i otu_table.tsv -o functional_table.tsv -g ../FAPROTAX.txt -c '#' -d 'taxonomy' --omit_columns 0 --column_names_are_in last_comment_line -r report_example1.txt -n columns_after_collapsing –v
collapse_table.py -i otu_table.biom -o functional_table.biom -g ../FAPROTAX.txt -r report_example2.txt -n columns_after_collapsing -v --collapse_by_metadata 'taxonomy'
4. BugBase
BugBase能进行表型预测,其中表型类型包括革兰氏阳性(Gram Positive)、革兰氏阴性(Gram Negative)、生物膜形成(Biofilm Forming)、致病性(Pathogenic)、移动元件(Mobile Element Containing)、氧需求(Oxygen Utilizing,包括Aerobic、Anaerobic、facultatively anaerobic)及氧化胁迫耐受(Oxidative Stress Tolerant)等7类。
脚本运行的例子
cd/home/shared/predict_pheno/BugBase_demo
run.bugbase.r -i HMP_s15.txt -m HMP_map.txt -c HMPBODYSUBSITE -o output
-i 基于Greengene数据库统计出的OTU丰度表
-m 映射文件
-o 输出的目录
5. FUNGuild
FUNGuild是一款基于ITS的功能预测软件。因为真菌基因组数据的缺乏,很难做KEGG通路的预测,所以FUNGuild是基于已发表的文章数据所整合起来的一种称为“guild”的分类预测。Guild是一种资源利用吸收所进行的功能分类,其中包括动物病原菌、植物病原菌、木质腐生菌等12类。通过对Guild的预测,可以对生态功能角度研究真菌的功能。
脚本运行例子
python Guilds_v1.0.py -otu /Users/username/Documents/project/otu_table_example.txt -db fungi -m -u
“
更多微生物组信息分析干货,
尽在锐翌基因培训班~
九月杭州,不见不散!
点击图片,获取培训班详情
参考文献
1. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature biotechnology. 2013;31(9):814–21. pmid:23975157; PubMed Central PMCID: PMC3819121.
2. Aßhauer KP, Wemheuer B, Daniel R, Meinicke P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics. 2015;31(17):2882–4.
3. Louca, S., Parfrey, L. W. & Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277 (2016)
4. Thomas A M, Jesus E C, Lopes A, et al. Tissue-associated bacterial alterations in rectal carcinoma patients revealed by 16S rRNA community profiling[J]. Frontiers in Cellular and Infection Microbiology, 2016, 6.
5. Toju H, Kishida O, Katayama N, et al. Networks Depicting the Fine-Scale Co-Occurrences of Fungi in Soil Horizons[J]. PloS one, 2016, 11(11): e0165987.
- 本文固定链接: https://oversea.maimengkong.com/kyjc/605.html
- 转载请注明: : 萌小白 2020年2月21日 于 卖萌控的博客 发表
- 百度已收录
