huanying今天给大家分享一个非常棒的玉米转录组的流程分析。原文作者是cxge,首发于omicshare论坛,阅读原文可跳转至本文的帖子哦~
软件及参考基因组
BWA, Samtools, Hisat2, HTseq, gffcompare, Stringtie, Ballgown,R
B73-V3, B73-V4(Enzampl database)
流程
protocol首先从原始RAN-seq数据入手,先经过质控fastqc,之后检测rRNA占比,去除杂的reads之后进行数据处理;使用HISAT2将读段匹配到参考基因组上,可提供注释文件;StringTie进行转录本组装,估算每个基因及isoform的表达水平;所有的转录本merge的数据再一次被呈递给StringTie,重新估算转录本的丰度,提供转录本reads数量的数据给下一步的ballgown;Ballgown从上一步获得所有转录本及其丰度,根据实验条件进行分类统计。
具体操作
1
质控检测
fastqc *.gz
Fail选项:
(1) Per Base Sequence Content(对所有reads的每一个位置,统计ATCG四种碱基正常情况的分布):当任一位置的A/T比例与G/C比例相差超过20%,报"FAIL"。
(2)Per Sequence GC Content:偏离理论分布的reads超过30%时,报"FAIL"。
(3)Duplicate
Sequences统计序列完全一样的reads的频率。测序深度越高,越容易产生一定程度的duplication,这是正常的现象,但如果duplication的程度很高,就提示我们可能有bias的存在(如建库过程中的PCR
duplication):当非unique的reads占总数的比例大于50%时,报"FAIL"
2
rRNA检测
NCBI网站下载Zea mays Eukaryotic 5S ribosomal RNA;Zea mays Eukaryotic 18S ribosomal RNA;Zea mays Eukaryotic 28S ribosomal RNA
# bwa index rRNA-maize.fa
# bwa mem -t 8 ../ref/rRNA-maize.fa NCSU_Wang_120430HiSeqRun_B73-WT- 22.fastq.gz | samtools view -bS - > B73-WT.bam
# samtools sort B73-WT.bam -o B73-WT.sort.bam
# samtools flagstat B73-WT.bam
3
玉米参考基因组及注释下载
# wget ftp://ftp.ensemblgenomes.org/pub/release-38/plants/fasta/ zea_mays/dna/Zea_mays.AGPv4.dna.toplevel.fa.gz
# wget ftp://ftp.ensemblgenomes.org/pub/release-31/plants/fasta/ zea_mays/dna/Zea_mays.AGPv3.31.dna.genome.fa.gz
# wget ftp://ftp.ensemblgenomes.org/pub/release-38/plants/gtf/ zea_mays/Zea_mays.AGPv4.38.gtf.gz
# wget ftp://ftp.ensemblgenomes.org/pub/release-31/plants/gtf/ zea_mays/Zea_mays.AGPv3.31.gtf.gz
从注释文件里面抽取出剪切位点和外显子信息,建立Index:
# extract_splice_sites.py Zea_mays.AGPv4.38.gtf > maize-v4.ss
#extract_exons.py Zea_mays.AGPv4.38.gtf > maize-v4.exon
# hisat2-build --ss maize-v4.ss --exon maize-v4.exon Zea_mays.AGPv4.dna.toplevel.fa maize-v4_tran 速度慢,限速
4
hisat2比对
#hisat2 -p 16 -x ./maize/Zea_mays_tran -U ./48/NCSU_Wang_120430HiSeqRun_B73-mt-48h.fastq.gz -S ./result/B73.sam
###多组数据用逗号隔开,双末端数据用-1,-2,单末端数据用-U,可以两种参数同时使用,reads长度可以不一致
# python -m HTSeq.s.count 统计reads number
5
组装转录本并定量表达基因
# stringtie B73.bam -p 8 -G
多个样品:
for i in *.sam;
do
i=${i%.sam*}; nohup samtools sort -@ 8 -o ${i}.bam ${i}.sam &
done
6
差异基因分析
R
>install.packages("devtools",repos="http://cran.us.r-project.org")
>source("http://www.bioconductor.org/biocLite.R")
>biocLite("ballgown", "RSkittleBrewer", "genefilter","dplyr","devtools")
>library(ballgown)
>library(RSkittleBrewer)
>library(genefilter)
>library(dplyr)
>library(devtools)
# vim phenodata.csv
ids, phenotype
sample-s1B73mt-22,mt
sample-s1B73WT-22,WT
sample-s2B73mt-22,mt
sample-s2B73WT-22,WT
sampleB73mt-22,mt
sampleB73WT-22,WT
# vim mergelistb73-22.txt
B73WT-22.gtf
B73mt-22.gtf
B73-WT-22.gtf
B73-mt-22.gtf
B73-WT-22.gtf
B73-mt-22.gtf
# stringtie --merge -p 8 –G Zea_mays.AGPv3.31.gtf -o b73_22.gtf mergelist_v3.txt
stringtie -e -B -p 8 -G b73_22.gtf –o sampleB73WT-22.gtf sampleB73WT-22.bam
stringtie -e -B -p 8 -G b73_22.gtf –o sampleB73mt-22.gtf sampleB73mt-22.bam
stringtie -e -B -p 8 -G b73_22.gtf –o sample-s1B73WT-22.gtf sample-s1B73WT-22.bam
stringtie -e -B -p 8 -G b73_22.gtf –o sample-s1B73mt-22.gtf sample-s1B73mt-22.bam
stringtie -e -B -p 8 -G b73_22.gtf –o sample-s2B73WT-22.gtf sample-s2B73WT-22.bam
stringtie -e -B -p 8 -G b73_22.gtf –o sample-s2B73mt-22.gtf sample-s2B73mt-22.bam
7
差异基因作图
7.1 导入数据
>pheno_data = read.csv("phenodata.csv")
Note: The
sample names you have stored in your phenotype file do not match the
file names of the samples you ran with StringTie!!!
> all(pheno_data$ids == list.files("ballgown"))
检查ballgown的list顺序:
list.files(“ballgown”)
bg = ballgown(dataDir = "ballgown",samplePattern = "sample",pData=pheno_data)
bg = ballgown(dataDir = "ballgown", samplePattern = "sample", meas = "all")
7.2滤掉低丰度的基因
>bg_filt = subset(bg,"rowVars(texpr(bg))>1",genomesubset=TRUE)
7.3确认组间有差异的转录本
>result_trans = stattest(bg_filt,feature="tran",covariate="phenotype",getFC=TRUE,meas="FPKM")
> result_trans
7.4确认组间有差异的基因
>result_gene = stattest(bg_filt,feature="gene",covariate="phenotype",getFC=TRUE,meas="FPKM")
> result_gene
7.5 对结果 result_trans增加基因名
>result_trans =data.frame(geneNames=ballgown::geneNames(bg_filt),geneIDs=ballgown::geneIDs(bg_filt), result_trans)
7.6按照P值排序(从小到大)
> result_trans=arrange(result_trans,pval)
> result_gene=arrange(result_gene,pval)
7.7把结果写入csv文件
> write.csv(result_trans, "tran_results.csv",row.names=FALSE)
> write.csv(result_gene, "gene_results.csv",row.names=FALSE)
7.8筛选出q值小于0.05的trans和genes
>subset(result_trans,result_trans$qval<0.05)
>subset(result_gene,result_gene$qval<0.05)
> write.csv(bg, "bg",row.names=FALSE)
8
数据可视化之颜色设定
# 赋予调色板五个指定颜色 > tropical= c('darkorange', 'dodgerblue','hotpink', 'limegreen', 'yellow') > palette(tropical)
# 当然rainbow()函数也可以完成这个任务 > palette(rainbow(5))
8.1以FPKM为参考值作图,以time作为区分条件
# 提取FPKM值
> fpkm = texpr(bg,meas="FPKM")
#方便作图将其log转换,+1是为了避免出现log2(0)的情况 > fpkm = log2(fpkm+1)
# 作图
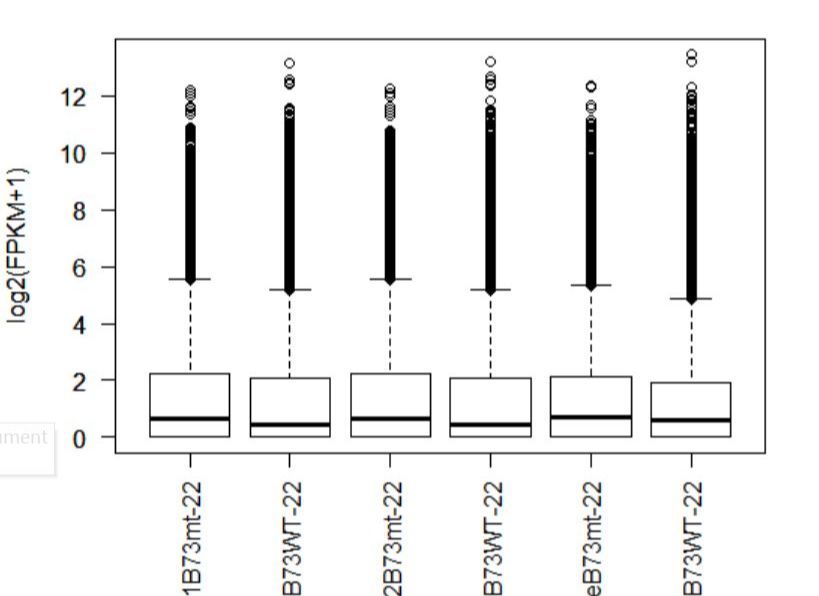
>boxplot(fpkm,col=as.numeric(pheno_data$sex),las=2,ylab='log2(FPKM+1)')
图1
8.2就单个转录本的查看在样品中的分布
> ballgown::tranNames(bg)[12]
12 "NR_027232"
> ballgown::geneNames(bg)[12]
12 "LINC00685"
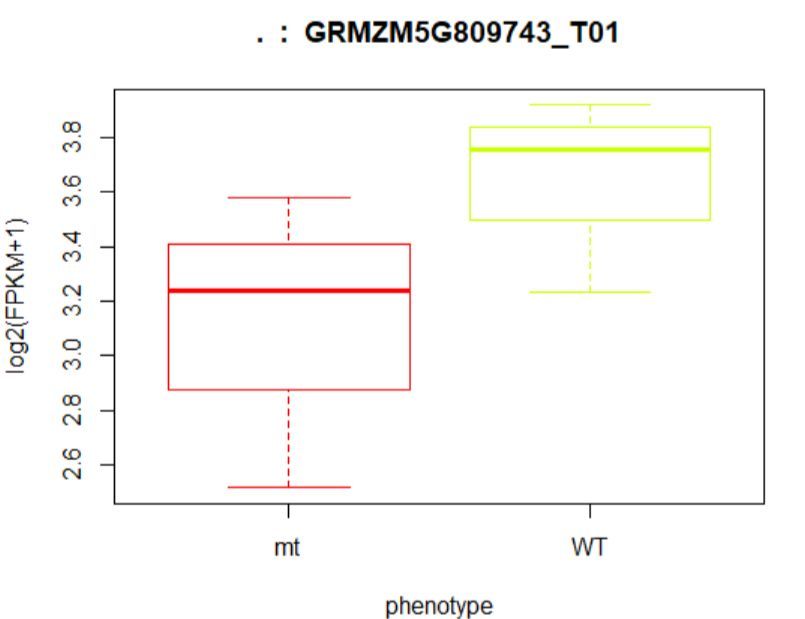
#绘制箱线图
> plot(fpkm[12,] ~ pheno_data$phenotype, border=c(1,2),
+ main=paste(ballgown::geneNames(bg)[12],' : ',
+ ballgown::tranNames(bg)[12]),pch=19, xlab="phenotype",
+ ylab='log2(FPKM+1)')
## (左图)
> points(fpkm[12,] ~ jitter(as.numeric(pheno_data$phenotype)),
+ col=as.numeric(pheno_data$phenotype))
## (右图)
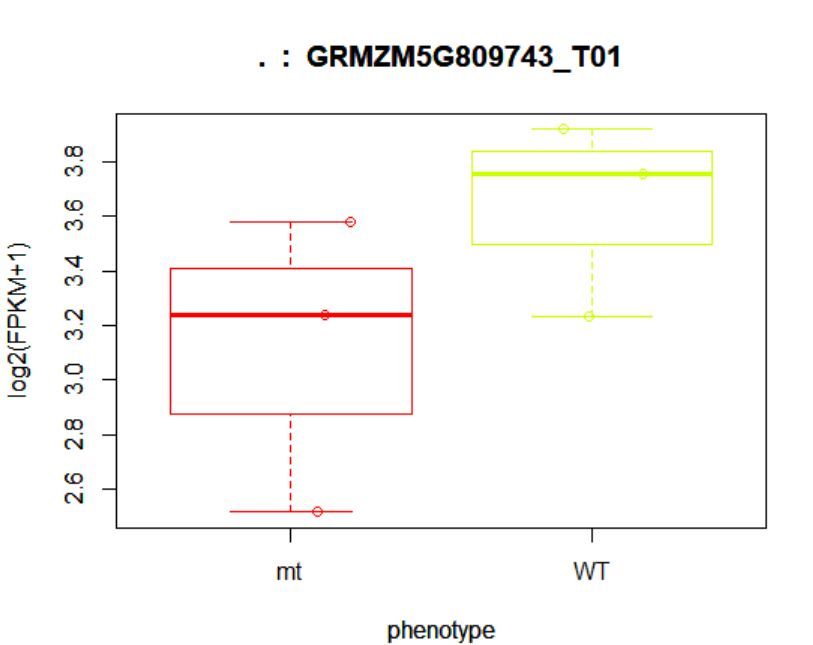
8.3查看某一基因位置上所有的转录本
>plotTrans(ballgown::geneIDs(bg)[442], bg, main=c('Gene MSTRG.168 in sample sampleB73WT-22'), sample=c('sampleB73WT-22'))
有空格就出错 图2
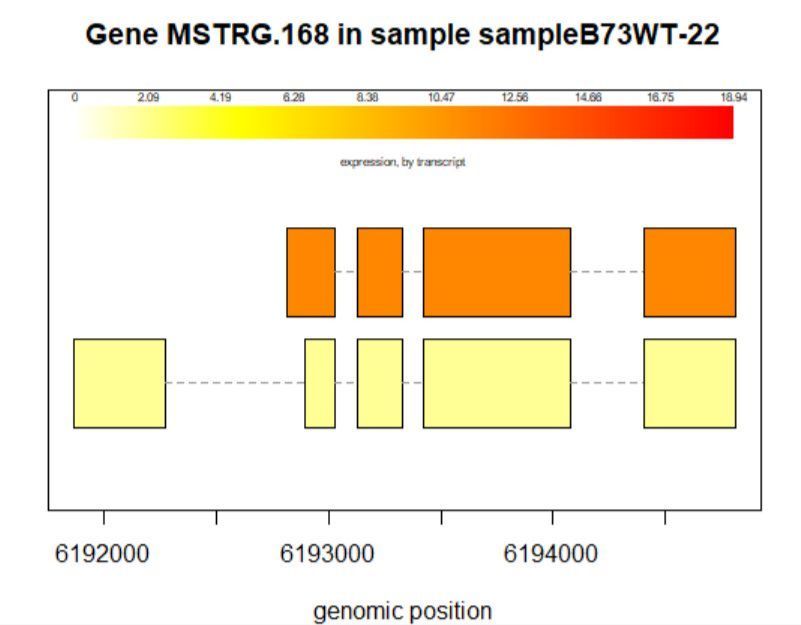
# plotTrans函数可以根据指定基因的id画出在特定区段的转录本
#可以通过sample函数指定看在某个样本中的表达情况,这里选用id=442, sample= sampleB73WT-22
> plotMeans('MSTRG.168',bg_filt,groupvar="phenotype",legend=FALSE)
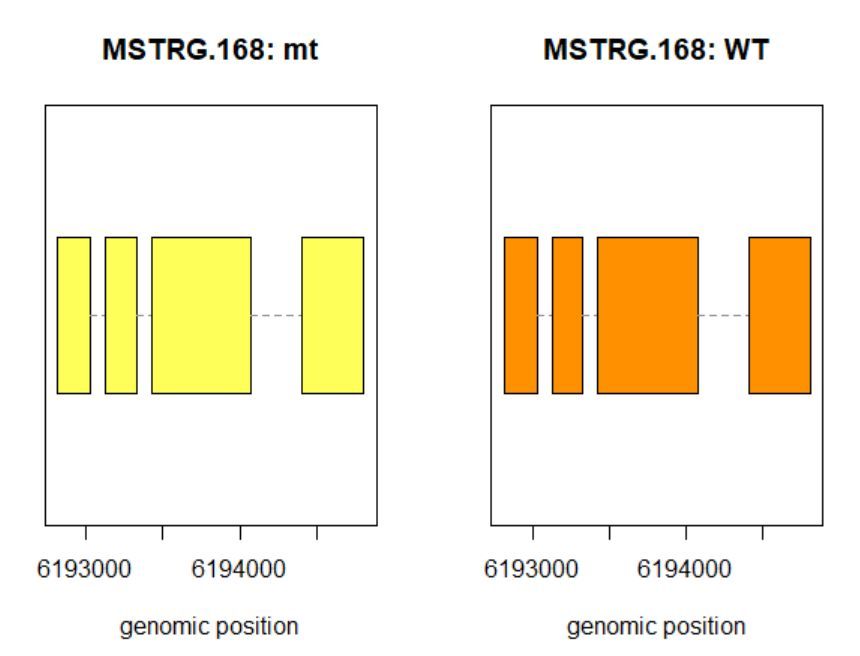
图3