RSeQC是一个RNA-Seq质控工具,提供了一系列有用的小工具能够评估高通量测序。其中一些基本模块:检查序列质量、核酸组分偏性、PCR偏性、GC含量偏性,还有RNA-seq特异性模块:评估测序饱和度、映射读数分布、覆盖均匀性、链特异性、转录水平RNA完整性等。下面我们就来介绍一下RSeQC的使用方法:
#安装RSeQC
tar zxf /opt/biosoft/RSeQC-2.6.4.tar.gz
cd RSeQC-2.6.4/
python setup.py install --root=/opt/bin/
export PYTHONPATH=/home/user/lib/python2.7/site-packages:$PYTHONPATH
export PATH=/opt/bin/usr/local/bin:$PATH
#分布区域计算
echo "read_distribution.py -i Col-16_1_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed 1>Col-16_1_unique.log
read_distribution.py -i Col-16_2_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed 1>Col-16_2_unique.log
read_distribution.py -i Col-16_3_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed 1>Col-16_3_unique.log
read_distribution.py -i mutant-16_1_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed 1>mutant-16_1_unique.log
read_distribution.py -i mutant-16_2_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed 1>mutant-16_2_unique.log
read_distribution.py -i
mutant-16_3_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed
1>mutant-16_3_unique.log" > command.read_distribution.list
sh command.read_distribution.list

统计了在外显子、内含子及非翻译区的情况
#统计reads在基因的分布
ls *.bam > bam_list.txt
geneBody_coverage.py -i bam_list.txt -r /opt/database/Arabidopsis/TAIR10.bed -o geneBody_coverage
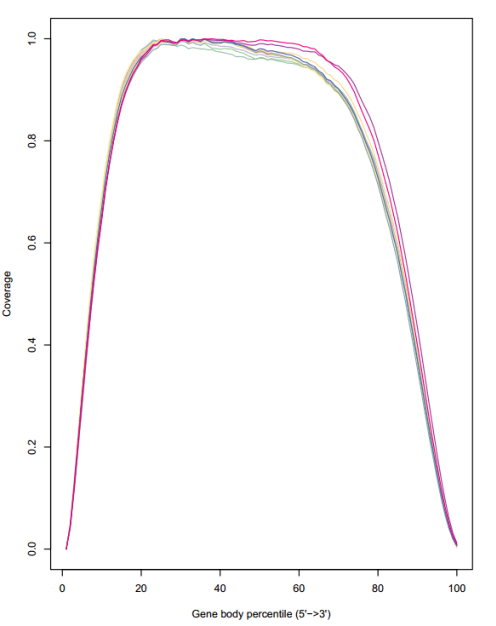

得到reads在基因上的折线图及热图
#新转录本统计
echo "junction_annotation.py -i Col-16_1_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed -o Col-16_1_junction_annotation
junction_annotation.py -i Col-16_2_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed -o Col-16_2_junction_annotation
junction_annotation.py -i Col-16_3_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed -o Col-16_3_junction_annotation
junction_annotation.py -i mutant-16_1_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed -o mutant-16_1_junction_annotation
junction_annotation.py -i mutant-16_2_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed -o mutant-16_2_junction_annotation
junction_annotation.py -i
mutant-16_3_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed -o
mutant-16_3_junction_annotation" > command.junction_annotation.list
sh command.junction_annotation.list
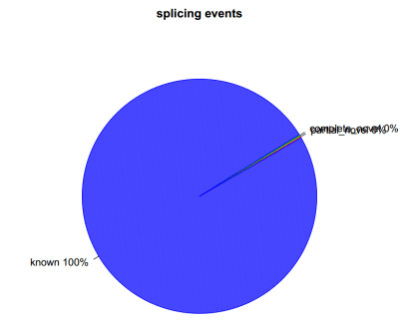
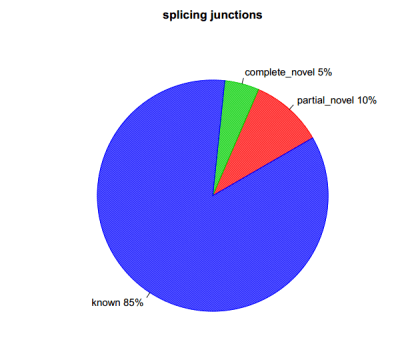
分别在剪切时间级别及剪切接头级别进行统计,分为与参考基因组完全一样,部分一样及完全不一样三种
#测序饱和度统计
echo "junction_saturation.py -i Col-16_1_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed -o Col-16_1_junction_saturation
junction_saturation.py -i Col-16_2_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed -o Col-16_2_junction_saturation
junction_saturation.py -i Col-16_3_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed -o Col-16_3_junction_saturation
junction_saturation.py -i mutant-16_1_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed -o mutant-16_1_junction_saturation
junction_saturation.py -i mutant-16_2_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed -o mutant-16_2_junction_saturation
junction_saturation.py -i
mutant-16_3_unique.bam -r /opt/database/Arabidopsis/TAIR10.bed -o
mutant-16_3_junction_saturation" > command.junction_saturation.list
sh command.junction_saturation.list
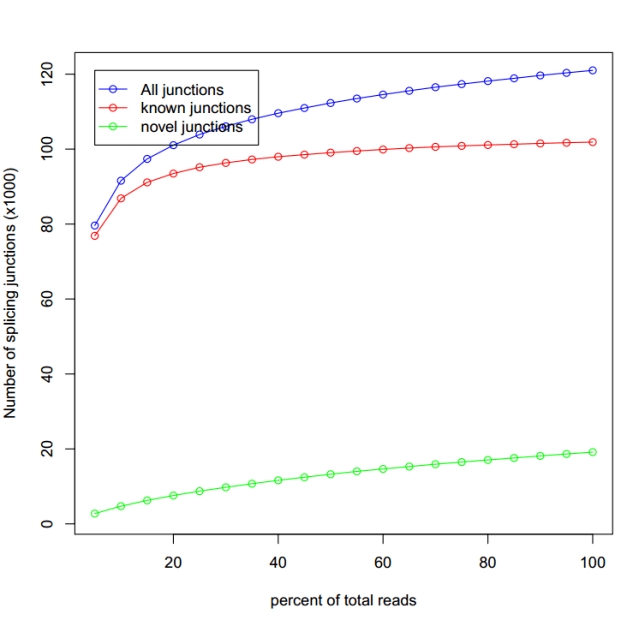
线趋于平就为饱和,表明测序深度够了
#不匹配统计
echo "mismatch_profile.py -l 125 -i Col-16_1_unique.bam -o Col-16_1_mismatch_profile
mismatch_profile.py -l 125 -i Col-16_2_unique.bam -o Col-16_2_mismatch_profile
mismatch_profile.py -l 125 -i Col-16_3_unique.bam -o Col-16_3_mismatch_profile
mismatch_profile.py -l 125 -i mutant-16_1_unique.bam -o mutant-16_1_mismatch_profile
mismatch_profile.py -l 125 -i mutant-16_2_unique.bam -o mutant-16_2_mismatch_profile
mismatch_profile.py -l 125 -i mutant-16_3_unique.bam -o mutant-16_3_mismatch_profile" > command.mismatch_profile.list
sh command.mismatch_profile.list
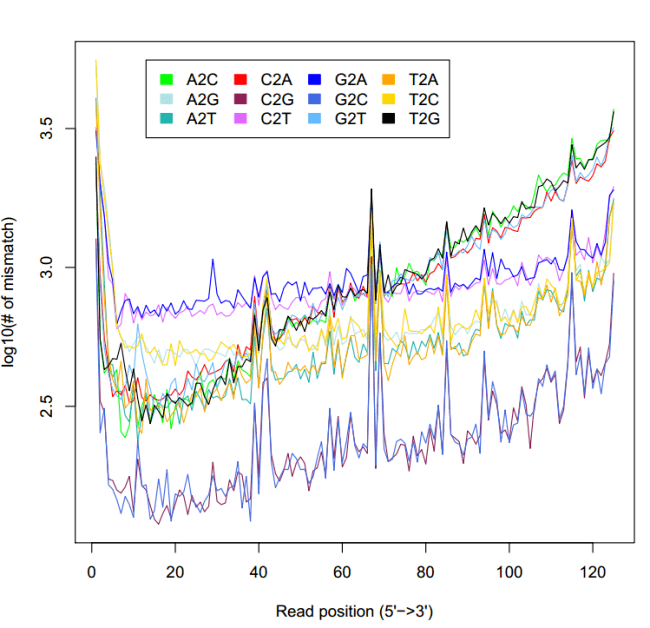
显示不匹配位点在reads位置的统计
#重复序列统计
echo "read_duplication.py -i Col-16_1_unique.bam -o Col-16_1_read_duplication
read_duplication.py -i Col-16_2_unique.bam -o Col-16_2_read_duplication
read_duplication.py -i Col-16_3_unique.bam -o Col-16_3_read_duplication
read_duplication.py -i mutant-16_1_unique.bam -o mutant-16_1_read_duplication
read_duplication.py -i mutant-16_2_unique.bam -o mutant-16_2_read_duplication
read_duplication.py -i mutant-16_3_unique.bam -o mutant-16_3_read_duplication" > command.read_duplication.list
sh command.read_duplication.list
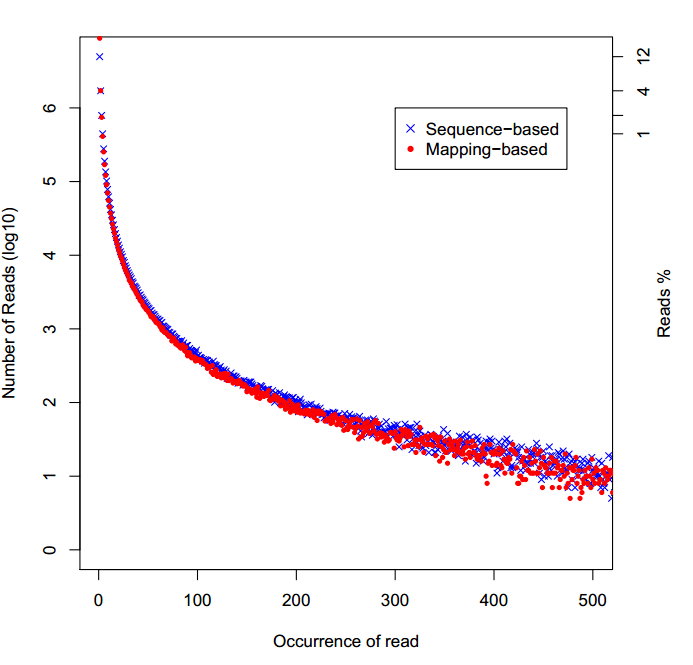
显示PCR重复序列的分布,一种是定义序列一样为重复序列,一种是定位map到同一位置的为重复序列
#GC含量统计
echo "read_GC.py -i Col-16_1_unique.bam -o Col-16_1_read_GC
read_GC.py -i Col-16_2_unique.bam -o Col-16_2_read_GC
read_GC.py -i Col-16_3_unique.bam -o Col-16_3_read_GC
read_GC.py -i mutant-16_1_unique.bam -o mutant-16_1_read_GC
read_GC.py -i mutant-16_2_unique.bam -o mutant-16_2_read_GC
read_GC.py -i mutant-16_3_unique.bam -o mutant-16_3_read_GC" > command.read_GC.list
sh command.read_GC.list
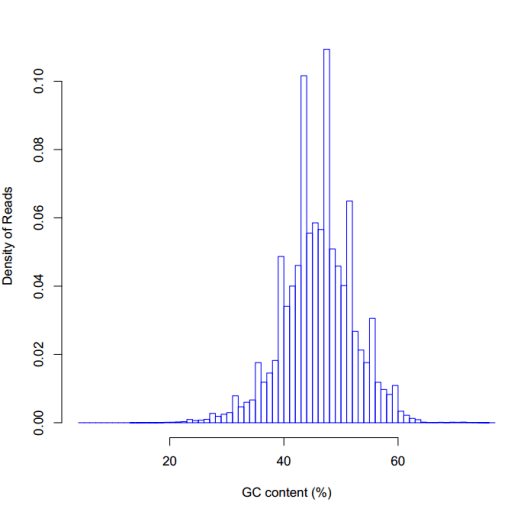
GC含量的分布
#计算插入片段大小
echo "inner_distance.py -i Col-16_1_unique.bam -o Col-16_1_inner_distance -r /opt/database/Arabidopsis/TAIR10.bed
inner_distance.py -i Col-16_2_unique.bam -o Col-16_2_inner_distance -r /opt/database/Arabidopsis/TAIR10.bed
inner_distance.py -i Col-16_3_unique.bam -o Col-16_3_inner_distance -r /opt/database/Arabidopsis/TAIR10.bed
inner_distance.py -i mutant-16_1_unique.bam -o mutant-16_1_inner_distance -r /opt/database/Arabidopsis/TAIR10.bed
inner_distance.py -i mutant-16_2_unique.bam -o mutant-16_2_inner_distance -r /opt/database/Arabidopsis/TAIR10.bed
inner_distance.py -i
mutant-16_3_unique.bam -o mutant-16_3_inner_distance -r
/opt/database/Arabidopsis/TAIR10.bed" > command.inner_distance.list
sh command.inner_distance.list
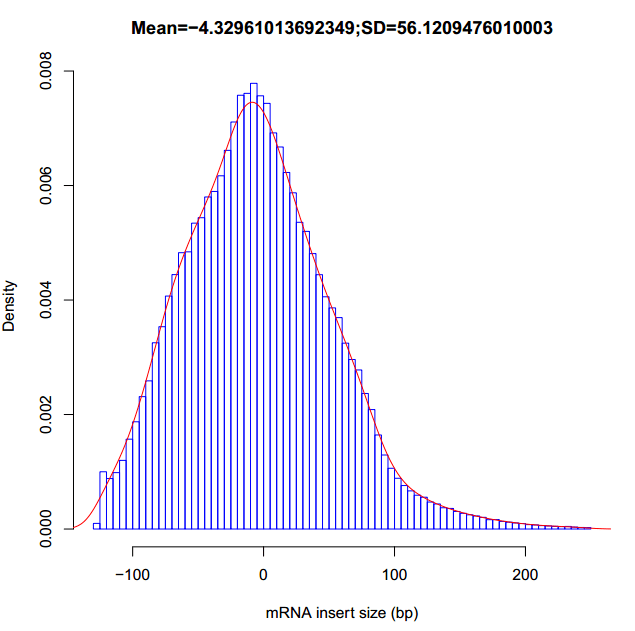
得到插入片段大小的平均值mean与标准偏差SD。