基于转录组测序获得的定量表达值,识别差异表达变化的基因或其它非编码RNA分子,实际上方法还是非常多的。但就目前来看,DESeq2和edgeR是出现频率最高的两种方法了。
DESeq2已经在前面文章中作了简介,本篇继续展示R包edgeR的差异基因分析流程。类似DESeq2,edgeR作为被广泛使用的R包,文献中经常能看到它的身影,如下举例。
 相关文献描述
相关文献描述
1 安装edgeR
edgeR可直接使用Bioconductor安装,还是非常简单的。
!!!**************************************************
#Bioconductor 安装 edgeR
#install.packages('BiocManager') #需要首先安装 BiocManager,如果尚未安装请先执行该步
BiocManager::install('edgeR')
!!!**************************************************
2 使用edgeR鉴定差异表达基因
edgeR使用经验贝叶斯估计和基于负二项模型的精确检验来确定差异基因,通过在基因之间来调节跨基因的过度离散程度,使用类似于Fisher精确检验但适应过度分散数据的精确检验用于评估每个基因的差异表达。
以下是edgeR分析差异表达基因的一般过程。
2.1 准备数据和文件读取
首先准备基因表达值矩阵。
本文的所有测试数据和R代码,可在文末获取
“control_treat.count.txt”,是6个测序样本的基因表达值矩阵,包括3个处理组(treat)和3个对照组(control),需注意将对照组在前,处理组在后。第1列是基因名称,注意不能有重复值。
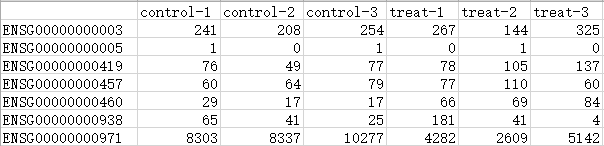 示例文件样式,行是基因,列是样本,内容为基因表达值
示例文件样式,行是基因,列是样本,内容为基因表达值
然后将准备好的基因表达值矩阵读到R中,并预定义样本分组信息。
!!!**************************************************
#读取基因表达矩阵
targets <- read.delim('control_treat.count.txt', row.names = 1, sep = '\t', check.names = FALSE)
#指定分组,注意要保证表达矩阵中的样本顺序和这里的分组顺序是一一对应的
#对照组在前,处理组在后
group <- rep(c('control', 'treat'), each = 3)
!!!**************************************************
2.2 表达值预处理和标准化
在执行差异表达基因分析前,将输入的基因表达矩阵和分组信息构建edgeR的DGEList对象,便于储存数据和中间运算;并对表达值标准化,以消除由于样品制备或建库测序过程中带来的影响。
同时,需要手动过滤一些低表达量的基因。可能会有同学疑问,为什么上一篇DESeq2中无需手动过滤呢?这是因为DESeq2设计之初就将自动过滤步骤封装在函数中,可以实现对低表达基因的自动处理;而edgeR则没有,因此需额外操作一下。推荐根据CPM(count-per-million,每百万碱基中目标基因的read
count值)值进行过滤,例如使用CPM值为1作为标准,即当某个基因在read count最低的样本(文库)中的count值大于(read
count最低的样品count总数/1000000),则保留。
!!!**************************************************
library(edgeR)
#数据预处理
#(1)构建 DGEList 对象
dgelist <- DGEList(counts = targets, group = group)
#(2)过滤 low count 数据,例如 CPM 标准化(推荐)
keep <- rowSums(cpm(dgelist) > 1 ) >= 2
dgelist <- dgelist[keep, , keep.lib.sizes = FALSE]
#(3)标准化,以 TMM 标准化为例
dgelist_norm <- calcNormFactors(dgelist, method = 'TMM')
!!!**************************************************
2.3 计算差异倍数列表
整体过程分为两步。
一是估计离散度。根据试验设计的样本分组,通过加权似然经验贝叶斯模型,估算每个基因表达的离散度,其代表了基因表达值在个体间的差异。
二是模型拟合。edgeR包中提供了多种计算差异基因的算法模型,之间略有不同,如下展示了两种。
!!!**************************************************
#差异表达基因分析
#首先根据分组信息构建试验设计矩阵,分组信息中一定要是对照组在前,处理组在后
design <- model.matrix(~group)
#(1)估算基因表达值的离散度
dge <- estimateDisp(dgelist_norm, design, robust = TRUE)
#(2)模型拟合,edgeR 提供了多种拟合算法
#负二项广义对数线性模型
fit <- glmFit(dge, design, robust = TRUE)
lrt <- topTags(glmLRT(fit), n = nrow(dgelist$counts))
write.table(lrt, 'control_treat.glmLRT.txt', sep = '\t', col.names = NA, quote = FALSE)
#拟似然负二项广义对数线性模型
fit <- glmQLFit(dge, design, robust = TRUE)
lrt <- topTags(glmQLFTest(fit), n = nrow(dgelist$counts))
write.table(lrt, 'control_treat.glmQLFit.txt', sep = '\t', col.names = NA, quote = FALSE)
!!!**************************************************
最后输出了各基因的差异表达倍数表。
以负二项广义对数线性模型的结果为例,包含了基因id、log2转化后的差异倍数(Fold Change)值、显著性p值以及校正后p值(默认FDR校正)等主要信息。
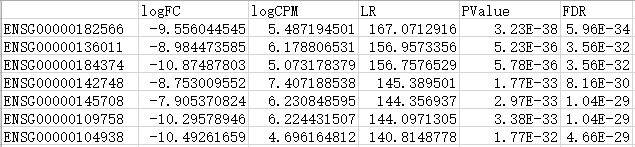 差异表达基因分析结果
差异表达基因分析结果
2.4 筛选差异表达基因
通过输出的差异表达倍数表,即可自定义阈值筛选差异表达基因了。
例如这里以上述输出的负二项广义对数线性模型的结果为例,将它再次读入到R中,并根据|log2FC|≥1以及FDR<0.01定义显著变化的基因,以及通过“up”和“down”分别区分上、下调的基因。
!!!**************************************************
##筛选差异表达基因
#读取上述输出的差异倍数计算结果
gene_diff <- read.delim('control_treat.glmLRT.txt', row.names = 1, sep = '\t', check.names = FALSE)
#首先对表格排个序,按 FDR 值升序排序,相同 FDR 值下继续按 log2FC 降序排序
gene_diff <- gene_diff[order(gene_diff$FDR, gene_diff$logFC, decreasing = c(FALSE, TRUE)), ]
#log2FC≥1 & FDR<0.01 标识 up,代表显著上调的基因
#log2FC≤-1 & FDR<0.01 标识 down,代表显著下调的基因
#其余标识 none,代表非差异的基因
gene_diff[which(gene_diff$logFC >= 1 & gene_diff$FDR < 0.01),'sig'] <- 'up'
gene_diff[which(gene_diff$logFC <= -1 & gene_diff$FDR < 0.01),'sig'] <- 'down'
gene_diff[which(abs(gene_diff$logFC) <= 1 | gene_diff$FDR >= 0.01),'sig'] <- 'none'
#输出选择的差异基因总表
gene_diff_select <- subset(gene_diff, sig %in% c('up', 'down'))
write.table(gene_diff_select, file = 'control_treat.glmQLFit.select.txt', sep = '\t', col.names = NA, quote = FALSE)
#根据 up 和 down 分开输出
gene_diff_up <- subset(gene_diff, sig == 'up')
gene_diff_down <- subset(gene_diff, sig == 'down')
write.table(gene_diff_up, file = 'control_treat.glmQLFit.up.txt', sep = '\t', col.names = NA, quote = FALSE)
write.table(gene_diff_down, file = 'control_treat.glmQLFit.down.txt', sep = '\t', col.names = NA, quote = FALSE)
!!!**************************************************
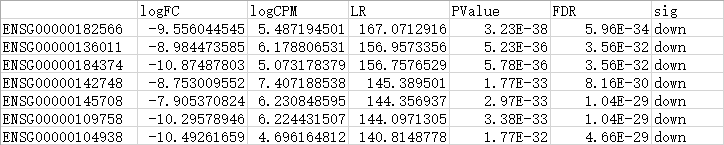 标识显著上下调的基因,添加在最后一列
标识显著上下调的基因,添加在最后一列
新获得的3个表格中,分别包含了根据自定义阈值筛选的所有差异基因、上调基因或下调基因列表,最后一列标注了上下调概况。
2.5 差异表达火山图示例
已经识别了显著差异表达的基因,最后期望通过火山图将它们表示出来,火山图是文献中常见统计图表之一。
ggplot2为此提供了优秀的作图方案,参考以下示例。
!!!**************************************************
##ggplot2 差异火山图
library(ggplot2)
#默认情况下,横轴展示 log2FoldChange,纵轴展示 -log10 转化后的 FDR
p <- ggplot(data = gene_diff, aes(x = logFC, y = -log10(FDR), color = sig)) +
geom_point(size = 1) + #绘制散点图
scale_color_manual(values = c('red', 'gray', 'green'), limits = c('up', 'none', 'down')) + #自定义点的颜色
labs(x = 'log2 Fold Change', y = '-log10 FDR', title = 'control vs treat', color = '') + #坐标轴标题
theme(plot.title = element_text(hjust = 0.5, size = 14), panel.grid = element_blank(), #背景色、网格线、图例等主题修改
panel.background = element_rect(color = 'black', fill = 'transparent'),
legend.key = element_rect(fill = 'transparent')) +
geom_vline(xintercept = c(-1, 1), lty = 3, color = 'black') + #添加阈值线
geom_hline(yintercept = 2, lty = 3, color = 'black') +
xlim(-12, 12) + ylim(0, 35) #定义刻度边界
p
!!!**************************************************
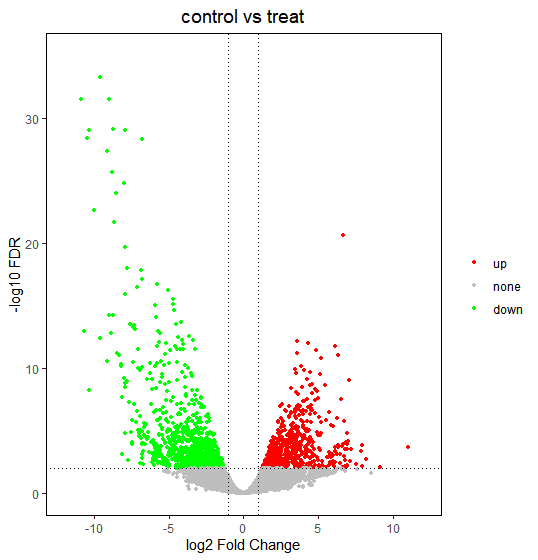 差异表达基因火山图
差异表达基因火山图
注:!!!*******之间为R脚本内容
转自:纪伟讲测序