大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
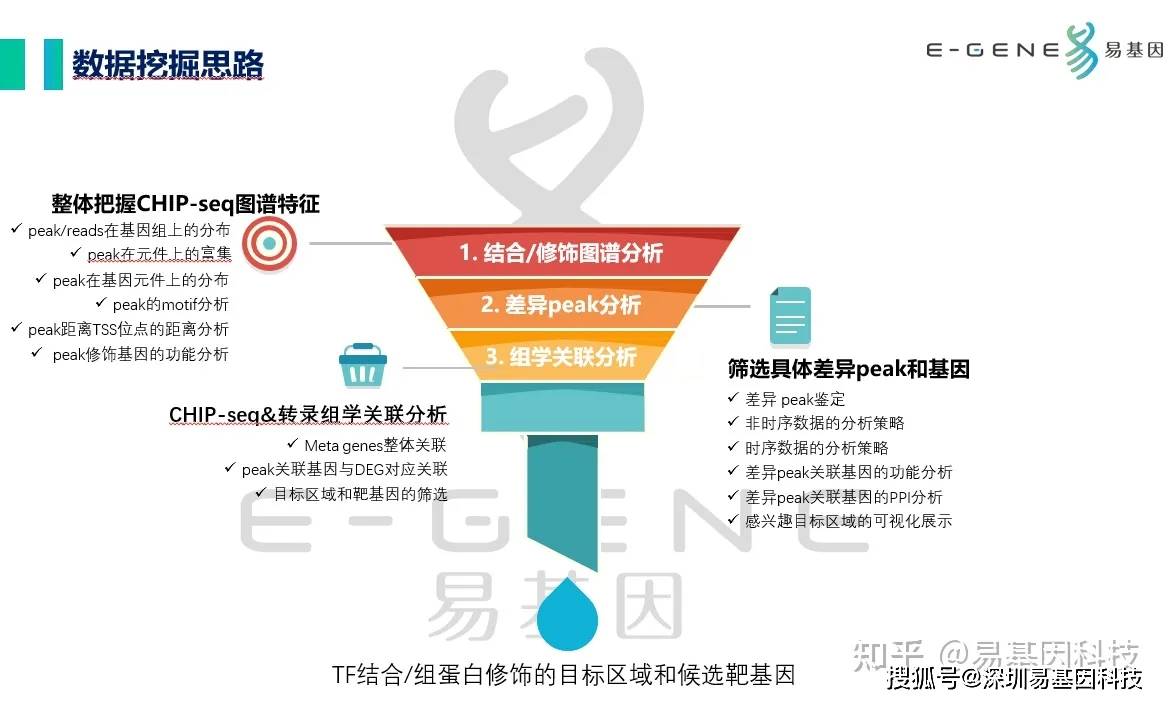
CHIP-seq研究的数据挖掘思路主要分为3步:
整体把握CHIP-seq图谱特征:peak/reads在基因组上的分布、peak在元件上的富集、peak在基因元件上的分布、peak的motif分析、peak距离TSS位点的距离分析、peak修饰基因的功能分析筛选具体差异peak和基因:差异
peak鉴定、非时序数据的分析策略、时序数据的分析策略、差异peak关联基因的功能分析、差异peak关联基因的PPI分析、感兴趣目标区域的可视化展示CHIP-seq&转录组学关联分析:Meta
genes整体关联、peak关联基因与DEG对应关联、目标区域和靶基因的筛选
后期视情况是否需要下游实验设计验证TF结合/组蛋白修饰的目标区域和候选靶基因。
1、图谱分析
(1)peak/reads在基因组上的分布
Peak的分布就是蛋白与DNA互作图谱。不同蛋白对DNA的结合可以按照峰的宽窄和分布特征分为:narrow
peak:即发生在DNA上特定的短序列,结合的区域很短。broad peak:这种类型的peak在DNA上呈弥
散的连续的分布,峰型较宽。一般来说,转录因子的峰型都是narrow peak;而对于组蛋白修饰,有的峰型为 narrow
peak,有的为broad peak。可以通过调整参数或使用不同的软件分别鉴定narrow peak及broad peak。

peak分布圈图
(2)信号的富集程度分析——覆盖度累积曲线
对样本比对结果reads累积情况进行展示。一定长度窗口(bin)上reads数进行计数,然后排序,再依次累加画图。input (能测到90
DNA片段)在基因组理论上是均匀分布,随着测序深度增加趋近于直线,实验组在排序越高的窗口处reads累积速度越快,说明这些区域富集的越特异。
narrow peak :富集程度高;broad peak:富集程度低。
富集程度低不代表失败, 如broad peak。但是如果是转录因子, 富集程度低则需要谨慎对待。
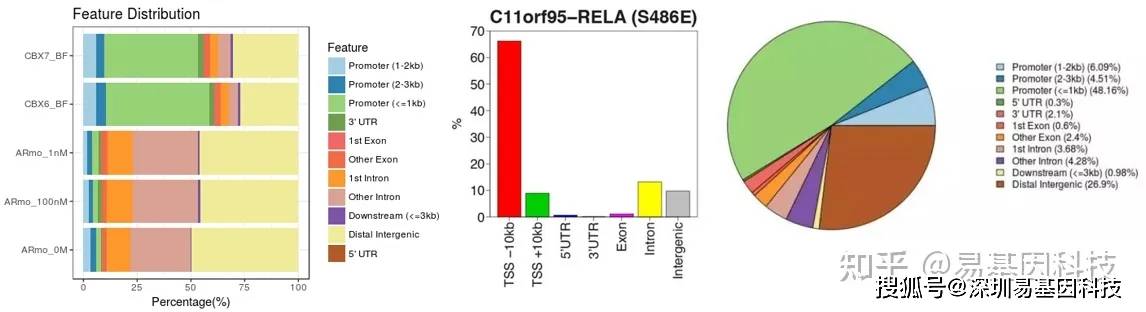
(3)peak/reads的基因元件富集分析
reference-point(relative to a point): 计算某个点的信号丰度scale-regions(over a
set of regions): 把所有基因组区段缩放至同样大小,然后计算其信号丰度。基于信号富集的靶基因集分类鉴定(基于聚类算法)
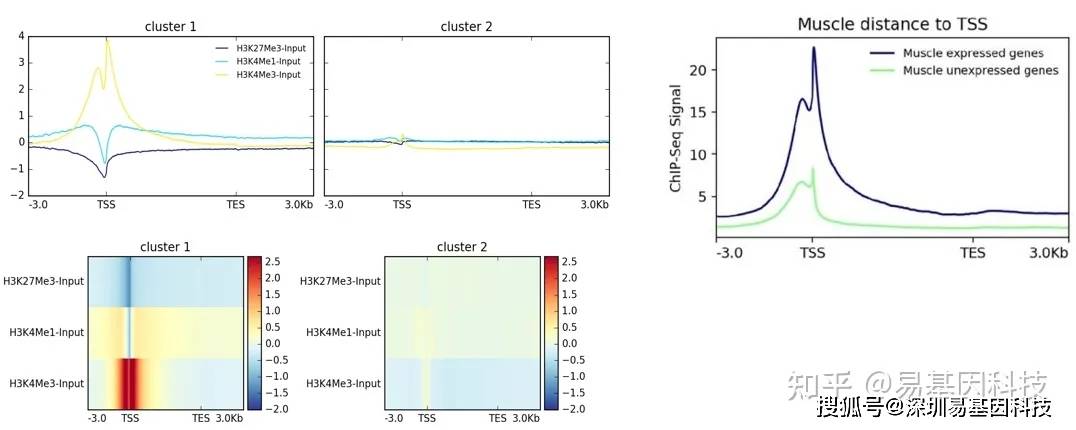
(4)peak/reads的基因元件分布分析
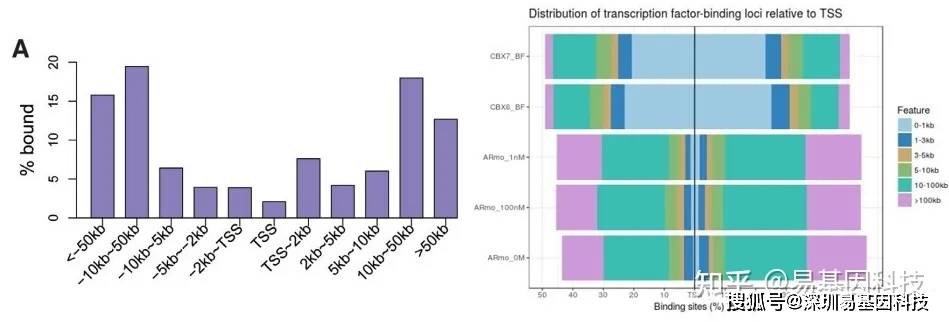
(5)peak/reads与TSS的相对距离分布
转录因子、组蛋白修饰往往具有重要的转录调控功能,而TSS附近是主要的转录调控区域,因此判断peak与TSS的位置关系有重要的意义。
(6)降维分析
将基因组分为等长窗口(bins),计算各样本各窗口内的Reads覆盖情况并进行标准化。基于此数据进行相关性、聚类和PCA分析。
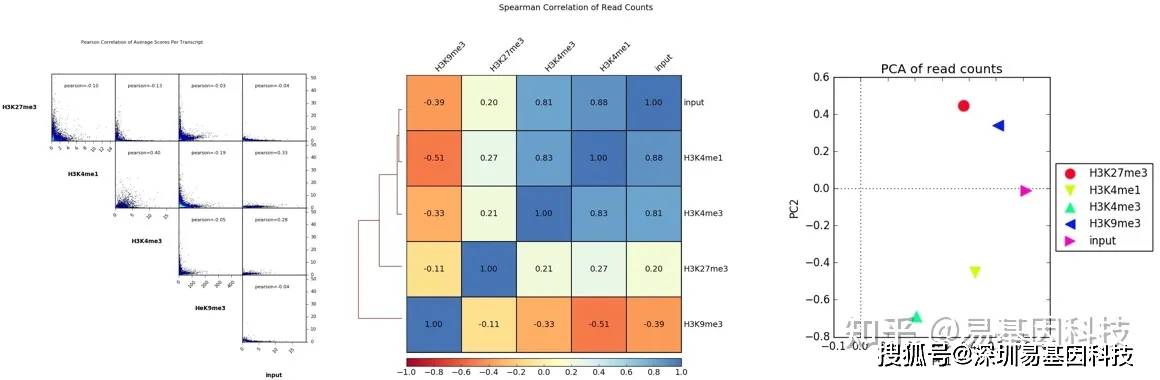
(7)motif分析
Motif为一段有特征的DNA短序列,主要为转录因子的识别位点,不同的motif对应不同的转录因子。
根据motif可以推测结合的转录因子。已知转录因子则分析该转录因子识别的序列特征。

(8)peak的基因注释和功能分析
ORAGSEA: 可以按照peak信号强度排序
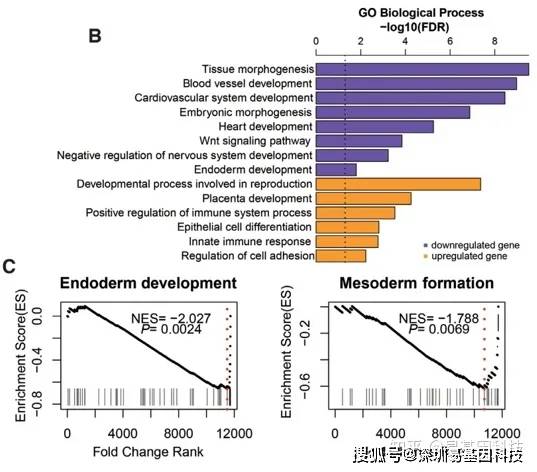
2、差异peak分析
(1)非时间序列数据:
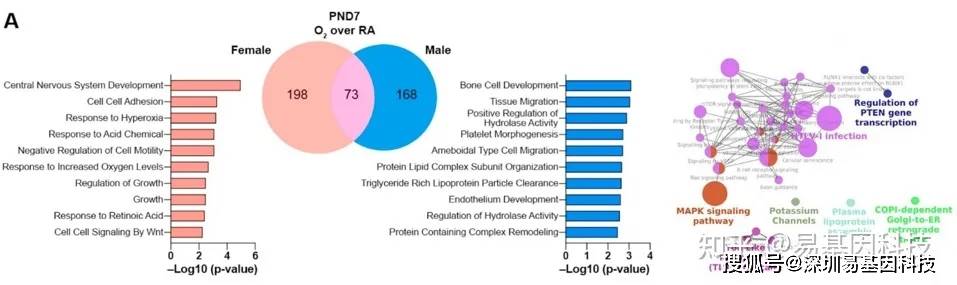
(2)时间序列数据:
(3)差异peak关联基因的PPI分析
感兴趣基因的差异peak展示
3、组学关联分析:CHIP-seq&转录组学
(1)Meta genes整体关联
距离TSS位点不同距离的peak注释到的基因的表达水平分析不同表达水平的基因,peak的数量分布对比
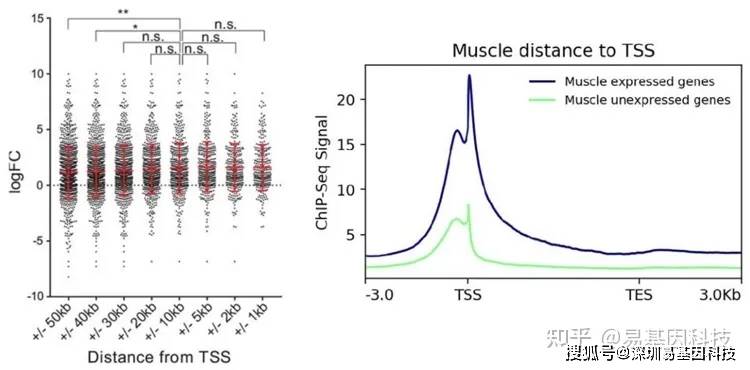
转录水平倍数变化 vs. peak倍数变化
(2)差异peak基因-DEG对应关联:筛选关键目的基因
peak关联基因与差异表达基因的重叠分析。peak关联基因可以是peak注释到启动子区,TSS±10kb区的基因,也可以来自已 知公共数据库的注释,如Human Enhancer Disease Database (HEDD)。九象限图法
关于易基因染色质免疫共沉淀测序 (ChIP-seq)
染色质免疫共沉淀(Chromatin
Immunoprecipitation,ChIP),是研究体内蛋白质与DNA相互作用的经典方法。将ChIP与高通量测序技术相结合的ChIP-Seq技术,可在全基因组范围对特定蛋白的DNA结合位点进行高效而准确的筛选与鉴定,为研究的深入开展打下基础。
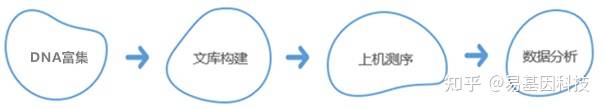
DNA与蛋白质的相互作用与基因的转录、染色质的空间构型和构象密切相关。运用组蛋白特定修饰的特异性抗体或DNA结合蛋白或转录因子特异性抗体富集与其结合的DNA片段,并进行纯化和文库构建,然后进行高通量测序,通过将获得的数据与参考基因组精确比对,研究人员可获得全基因组范围内某种修饰类型的特定组蛋白或转录因子与基因组DNA序列之间的关系,也可对多个样品进行差异比较。
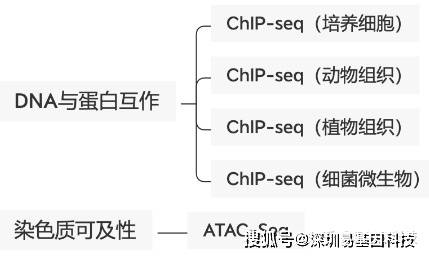
应用方向:
ChIP 用来在空间上和时间上不同蛋白沿基因或基因组定位
转录因子和辅因子结合作用复制因子和 DNA 修复蛋白组蛋白修饰和变异组蛋白
技术优势:
物种范围广:细胞、动物组织、植物组织、细菌微生物多物种富集经验;微量建库:只需5ng以上免疫沉淀后的DNA,即可展开测序分析;方案灵活:根据不同的项目需求,选择不同的组蛋白修饰特异性抗体。
技术路线:
有ChIP-seq测序或组学研究需要的老师可联系易基因。

转自:深圳易基因