这一步主要看看这些外显子测序数据的测序质量如何:
首先用fastqc处理,会出一些图表,肯定是没问题的啦,如果数据有问题,公司就不会给你,那样不砸了他们自己的招牌嘛。
然后我们粗略统计下平均测序深度及目标区域覆盖度,这个是重点,不过一般没问题的,因为现在芯片捕获技术非常成熟了,而且实验水平大幅提升,没有以前那么多的问题了。
这个外显子项目的测序文件里面,mpileup文件是1371416525行,意味着总的测序长度是1.3G,以前我接触的一般是600M左右的
因为外显子目标区域并不大,就34729283bp,也就是约35M。
即使加上侧翼长度
54692160 外显子加上前后50bp
73066288 外显子加上前后100bp
90362533 外显子加上前后150bp
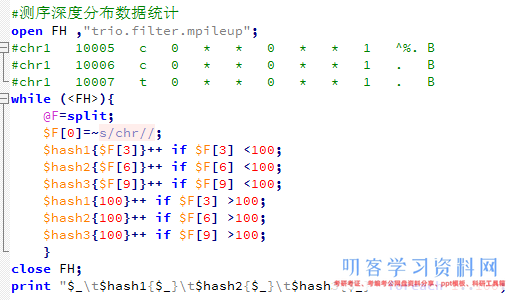
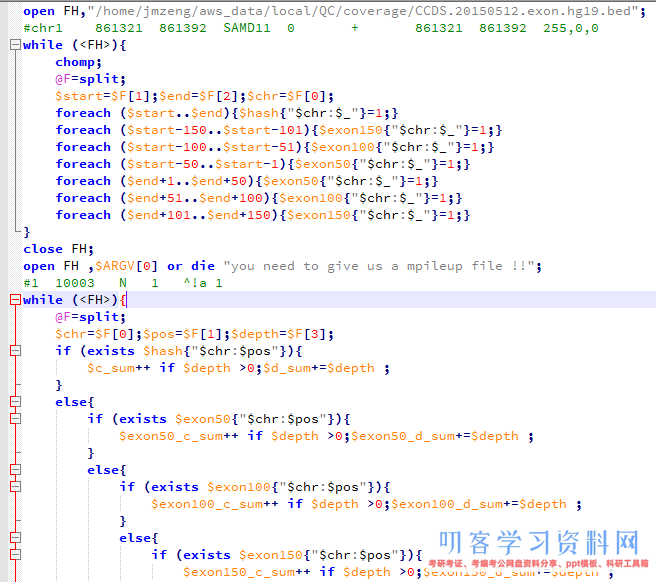
然后我要根据外显子记录文件对mpileup文件进行计数,统计外显子coverage,还有测序深度,这个脚本其实蛮有难度的!
我前面提到过外显子组的序列仅占全基因组序列的1%左右,而我在NCBI里面拿到 consensus coding sequence
(CCDS)记录CCDS.20150512.txt文件,是基于hg19版本的,需要首先转换成hg18才可以来计算这次测序项目的覆盖度和平均测序深度。
参考: liftover基因组版本之间的coordinate转换
awk ‘{print “chr”$3,$4,$5,$1,0,$2,$4,$5,”255,0,0″}’ CCDS.20150512.exon.txt >CCDS.20150512.exon.hg38.bed
~/bio-soft/liftover/liftOver CCDS.20150512.exon.hg38.bed
~/bio-soft/liftover/hg38ToHg19.over.chain CCDS.20150512.exon.hg19.bed
unmap
下面这个程序就是读取转换好的外显子记录的数据,对一家三口一起统计,然后再读取每个样本的20G左右的mpileup文件,进行统计,所以很耗费时间。
外显子目标区域平均测序深度接近100X,所以很明显是非常好的捕获效率啦!而全基因组背景深度才3.3,这 符合实验原理, 即与探针杂交碱基多的片段比少的片段更易被捕获. 对非特异杂交的,基因组覆盖度非特异的背景 DNA 也进行了测序。
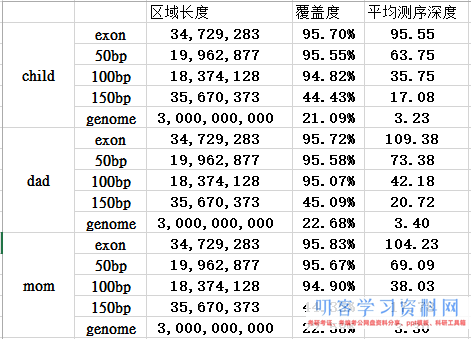
接下来对测序深度进行简单统计,脚本如下,但是这个图没多大意思。因为我们的外显子的35M区域平均都接近100X的测序量