理解ChIP-Seq
到了目前这个水平,我学习新的高通量数据分析流程时已经不再考虑代码应该如何写的问题了。我更多要去考虑一个技术的目的和意义。
转录组主要研究的问题是基因在不同情况下的差异表达以及RNA结构变化等,而表观组研究的问题是在基因序列不变的情况下,基因的表达、调控和性状发生了可遗传变化的分子机制。也就是相同的DNA, RNA,蛋白质经过一定的修饰后会使得生物性状发生了改变。
ChIP-Seq仅仅是第一个表观遗传学领域比较成熟的技术而已,目前还有很多其他的技术,比如说
DNA修饰: DNA甲基化免疫共沉淀技术(MeDIP), 目标区域甲基化,全基因组甲基化(WGBS),氧化-重亚硫酸盐测序(oxBS-Seq), TET辅助重亚硫酸盐测序(TAB-Seq)
RNA修饰: RNA甲基化免疫共沉淀技术(MeRIP)
蛋白质与核酸相互作用: RIP-Seq, ChIP-Seq, CLIP-Seq
还有最近比较火的ATAC-Seq
2013年PNAS上面有一篇文章叫做 Epigenetics: Core misconcept 讲了几点关于表观遗传学,大家的理解错误的地方。
本次主要是分析ChIP-Seq的高通量测序结果,因此,先介绍什么是ChIP-Seq.
所谓的ChIP-Seq其实就是把ChIP实验做完得到的DNA不仅仅用来跑胶,还送去高通量测序了。重点在于ChIP,也就是染色体免疫共沉淀(Chromatin
Immunoprecipitation)是用来解决什么科学问题的。毕竟数据分析不是目的,它只是解决问题的一种手段。
ChIP被用来研究细胞内DNA与蛋白质相互作用,具体来说就是确定特定蛋白(如转录因子)是否结合特定基因组区域(如启动子或其它DNA结合位点)——可能定义顺反组。ChIP还被用来确定基因组上与组蛋白修饰相关的特定位点(即组蛋白修饰酶类的靶标)
- 来自维基百科。
ChIP的实验我从来没有做过,所以只能从网上找来大致的流程,简单分为
第一步: 将蛋白交联到DNA上。 也就是保证蛋白和DNA能够结合,找到互作位点。
第二步: 通过超声波剪切DNA链。
第三步: 加上附上抗体的磁珠用于免疫沉淀靶蛋白。抗体很重要
第四步: 接触蛋白交联;纯化DNA
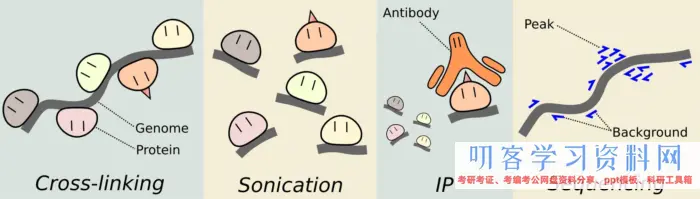
实验流程
如果送去测序就是ChIP-Seq, 后续就是对应分析流程,分析分为4步:
质量控制, 用到的是FastQC
序列比对,Bowtie2或这BWA
peak calling, 建议用MACS
peak注释, 推荐Y叔的ChIPseeker
文章中ChIP-Seq解决的问题
这部分看看这篇文章想解决什么问题,以及ChIP-Seq解决了什么问题。工具一定要为目的服务。
PS: 可以先去把数据下载了,再看这个部分
文章的研究内容:
PRC1(Polycomb repressive complex 1)与干细胞命运决定有关。PRC1的组成非常的复杂,原文这样写道:
The
protein families that constitute the core of PRC1 contain several
members: Cbx (Cbx2, Cbx4, Cbx6, Cbx7, or Cbx8); Ring1A or Ring1B; PHC
(PHC1, PHC2, or PHC3); PCGF (PCGF1, PCGF2, PCGF3, PCGF4, PCGF5, or
PCGF6); and RYBP or YAF2. Each combination establishes the subtype of
PRC1 complexes
文章想解决的问题是:
PRC1复合体两类亚型的全基因组定位
两类复合体调节的基因表达是否有差异
Cbx7,RYBP是否有共同或独特的生物学功能
Cbx7,RYBP在染色质的定位是否是相互依赖
得到的部分答案是:小鼠中有两个互斥的PRC1复合体的变异体,Cbx7和RYBP。Cbx7和RYBP的在基因组上的结合在同一个位置,但是同样互斥,不能共存。
Cbx7用于招募Ring1B结合到染色质,RYBP增加PRC1酶活性。
RYBP所结合的基因,有着较低水平Ring1B和H2AK119ub的水平,表达量高于Cbx7结合态。RYBP和Cbx7的在基因位点的结合情况还与代谢调节,细胞周期过程有关。
ChIP的用途:
为了解决PRC1亚型的定位问题, 作者对PRC1复合体的组成Cbx7, Ring1B, RYBP, 以及PRC2的Suz12f分别做了ChIP-Seq. 然后就是画了很多韦恩图看不同蛋白的靶基因的相互关系。还看了不同蛋白在基因的上的位置。
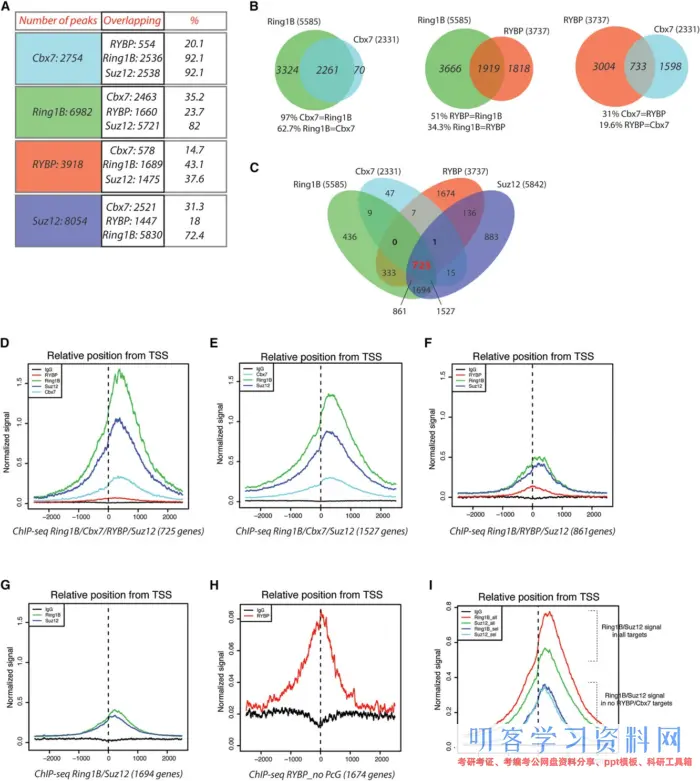
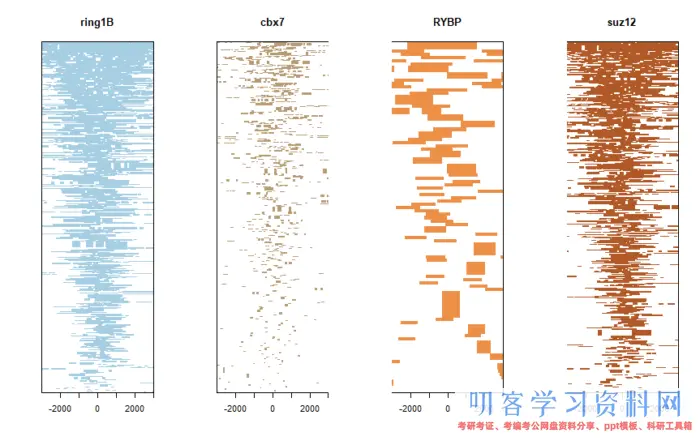
一般而言,ChIP-Seq基本就是得到上面这些图,根据疑问再找到某一些基因看调控蛋白的结合和定位。本次实战也就是尝试做出这些图。
根据原文,ChIP-Seq的流程如下:
先用Bowtie(0.12.7)把RYBP, Cbx7, Ring1B, Suz12,和H2AK11Ub的ChIP-Seq产生短读(reads) 比对到小鼠的参考基因组上(NCBIM37),参数允许seed联配时有2个错配。
使用MACS(1.4.1)寻找可能的结合位点,也就是基因组中大量短读片段富集的区域。MACS比较这些区域在对照组和实验组的差异,选择p值等于10e-5作为阈值。
之后PDR低于5%的Cbx7, Ring1B, Suz12和H2AK119ub的peak和PDR低于1%的RYBP的peak结合用于进一步分析
这一步看起来就很复杂,之后会重点进行学习
在基因内部或距离TSS
2.5kb的 peak 被认为是靶基因。 每个TSS附近5Kb区域,并且与一个或多个ChIP-Seq(RYBP,Ring1B, Cbx7,
Suz12, H2AK119ub)结果关联,用于计算ChIP-Seq表达谱和全ChIP-Seq覆盖度。
使用BEDtools 计算TSS附近ChIP-Seq表达谱
ChIP-Seq数据分析的流程难点在于找到peak,所以peak calling这一阶段的软件选择比较重要,目前常用的是MACS2, 实际分析建议多用几个软件。目前常用的是MACS2, 实际分析建议多用几个软件。
数据下载和质量控制
这部分下载ChIP-Seq数据,以及小鼠的参考基因组,注释。
下载测序数据,并用sratoolkit解压缩文章提供的GEO是GSE42466,可以在https://www.ncbi.nlm.nih.gov/geo/ 检索到对应的数据集

GEO
# 我习惯将数据保存到项目文件夹下的data中
mkdir -p GSE42466/data/chip-seq
cd GSE42466/data/chip-seq
for ((i=204;i<=209;i++));
do
wget -4 -q ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP017/SRP017311/SRR620$i/SRR620$i.sra;
~/miniconda/envs/biostar/bin/fastq-dump –split-3 SRR620$i.sra;
done &
下载小鼠参考基因组的索引和注释文件, 这里用常用的mm10
# 索引大小为3.2GB, 不建议自己下载基因组构建
mkdir referece && cd reference
wget -4 -q ftp://ftp.ccb.jhu.edu/pub/data/bowtie2_indexes/mm10.zip
unzip mm10.zip
注释文件到https://genome.ucsc.edu/cgi-bin/hgTables 选取保存。
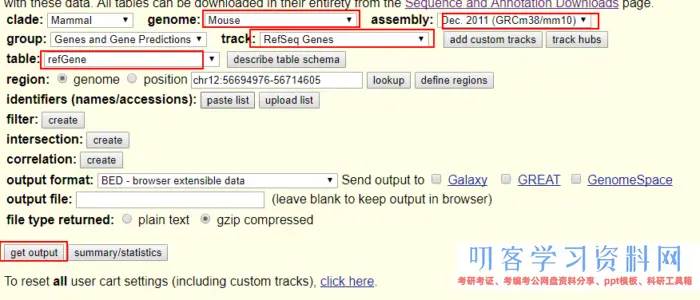
或者用curl进行下载。
curl \'https://genome.ucsc.edu/cgi-bin/hgTables?hgsid=603641571_MKLxB78UAcjaI3oj5YiFCA0Ms86o&boolshad.hgta_printCustomTrackHeaders=0&hgta_ctName=tb_refGene&hgta_ctDesc=table+browser+query+on+refGene&hgta_ctVis=pack&hgta_ctUrl=&fbQual=whole&fbUpBases=200&fbExonBases=0&fbIntronBases=0&fbDownBases=200&hgta_doGetBed=get+BED\' > mm10_refSeq.bed
得到的fastq文件,一般而言都是上机得到的原始数据,也有可能是经过质量控制的结果,但是无论如何都得自己去检查一下。
## 需要安装fastqc 和 multiqc
# 先获取QC结果
ls *gz | while read id; do fastqc -t 4 $id; done
# multiqc
multiqc *fastqc.zip --pdf
根据质控结果,发现序列的前后几个碱基质量不好,因此在比对的时候会主动忽略。
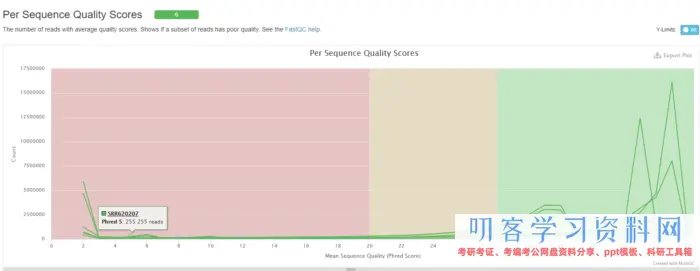
今后,可能会去下载其他实验的数据,还要进行质量控制,所以就写一个Python脚本进行流程化操作。脚本命名为sra2qc.py,保存在我的GitHub上
序列比对
这一步非常简单, 就是将得到fastq文件用bowtie2比对小鼠参考基因组上,
bowtie2 -p 6 -3 5 --local -x reference/mm10 -U ChIP-Seq/SRR620204.fastq | ~/miniconda3/bin/samtools sort -O bam -o ../analysis/alignment/ring1B.bam
bowtie2 -p 6 -3 5 --local -x reference/mm10 -U ChIP-Seq/SRR620205.fastq | ~/miniconda3/bin/samtools sort -O bam -o ../analysis/alignment/cbx7.bam
bowtie2 -p 6 -3 5 --local -x reference/mm10 -U ChIP-Seq/SRR620206.fastq | ~/miniconda3/bin/samtools sort -O bam -o ../analysis/alignment/suz12.bam
bowtie2 -p 6 -3 5 --local -x reference/mm10 -U ChIP-Seq/SRR620207.fastq | ~/miniconda3/bin/samtools sort -O bam -o ../analysis/alignment/RYBP.bam
bowtie2 -p 6 -3 5 --local -x reference/mm10 -U ChIP-Seq/SRR620208.fastq | ~/miniconda3/bin/samtools sort -O bam -o ../analysis/alignment/IgGold.bam
bowtie2 -p 6 -3 5 --local -x reference/mm10 -U ChIP-Seq/SRR620209.fastq | ~/miniconda3/bin/samtools sort -O bam -o ../analysis/alignment/IgG.bam
我对比对结果进行了简单的统计,发现比对率其实不太高. 虽然在RNA-Seq是要求80~90%的比对率,但是在ChIP-Seq上,我没有具体了解过。
样品 | 比对到单个区域 | 比对到多个区域 |
|---|---|---|
ring1B | 43.76% | 15.81% |
cbx7 | 39.39% | 17.49% |
suz12 | 48.88% | 17.17% |
RYBP | 58.76% | 28.12% |
IgGold | 54.54% | 27.21% |
IgG | 57.83% | 25.45% |
比对的结果,可以打开IGV查看,和RNA-Seq最大的区别在于你能看到明显的峰,这就是peak。这类peak在全基因组上特别的多,我们无法用肉眼直接选择,因此就需要借助专门的工具查找。
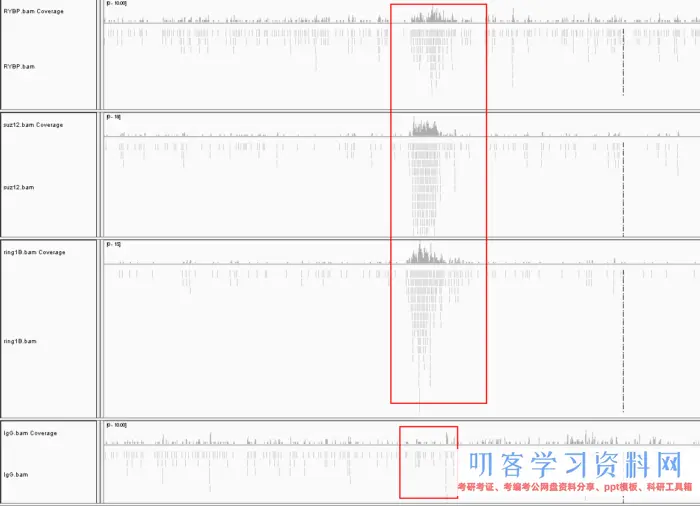
一般ChIP-Seq都需要提供一个对照组,也就是图中IgG。很明显的发现其他实验组有峰的区域在对照组中是没有的。而实验组没有峰的区域,对照组也没有,这样子就能提出背景噪声。
peak calling
peak calling要用到MACS, 建议用ananconda安装, 因为可以选择合适的Python环境。
conda create -n macs python=2
source activate macs
pip install MACS2
macs2包含一系列的子命令,其中最主要的就是callpeak, 官方提供了使用实例
macs2 callpeak -t ChIP.bam -c Control.bam -f BAM -g hs -n test -B -q 0.01
一般而言,我们照葫芦画瓢,按照这个实例替换对应部分就行了,介绍一下各个参数的意义
-t: 实验组的输出结果
-c: 对照组的输出结果
-f:
-t和-c提供文件的格式,可以是\"ELAND\", \"BED\", \"ELANDMULTI\", \"ELANDEXPORT\",
\"ELANDMULTIPET\" (for pair-end tags), \"SAM\", \"BAM\", \"BOWTIE\",
\"BAMPE\" \"BEDPE\" 任意一个。如果不提供这项,就是自动检测选择。
-g: 基因组大小, 默认提供了hs, mm, ce, dm选项, 不在其中的话,比如说拟南芥,就需要自己提供了。
-n: 输出文件的前缀名
-B: 会保存更多的信息在bedGraph文件中,如fragment pileup, control lambda, -log10pvalue and -log10qvalue scores
-q: q值,也就是最小的PDR阈值, 默认是0.05。q值是根据p值利用BH计算,也就是多重试验矫正后的结果。
-p: 这个是p值,指定p值后MACS2就不会用q值了。
-m: 和MFOLD有关,而MFOLD和MACS预构建模型有关,默认是5:50,MACS会先寻找100多个peak区构建模型,一般不用改,因为你不懂。
原文写到:MACS
estimated the FDR by comparing the peaks obtained from the samples with
those from the control samples, using the same p value cutoff (10e-5).
估计就是指文章用了q=0.05或0.01
macs2 callpeak -c IgGold.bam -t suz12.bam -q 0.05 -f BAM -g mm -n suz12 2> suz12.macs2.log &
macs2 callpeak -c IgGold.bam -t ring1B.bam -q 0.05 -f BAM -g mm -n ring1B 2> ring1B.macs2.log &
macs2 callpeak -c IgG.bam -t cbx7.bam -q 0.05 -f BAM -g mm -n cbx7 2> cbx7.macs2.log &
macs2 callpeak -c IgG.bam -t RYBP.bam -q 0.01 -f BAM -g mm -n RYBP 2>RYBP.macs2.log &
可以看下找到了多个peak,
wc -l *bed
2384 cbx7_summits.bed
8342 ring1B_summits.bed
0 RYBP_summits.bed
7619 suz12_summits.bed
结果显示RYBP无peak,估计是数据上传错误,我们可以下载作者上传的peak数据。此外ring1B我找了8000多个,但是原文是7000个,或许要修改阈值。
wget ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE42nnn/GSE42466/suppl/GSE42466_Suz12_peaks_10.txt.gz
gzip -d GSE42466_Suz12_peaks_10.txt.gz
mv GSE42466_RYBP_peaks_5.txt RYBP2_summits.bed
结果文件不只是上述提到的一类,还有如下格式:
NAME_peaks.xls: 以表格形式存放peak信息,虽然后缀是xls,但其实能用文本编辑器打开,和bed格式类似,但是以1为基,而bed文件是以0为基.也就是说xls的坐标都要减一才是bed文件的坐标
NAME_peaks.narrowPeak
NAME_peaks.broadPeak 类似。后面4列表示为, integer score for display,
fold-change,-log10pvalue,-log10qvalue,relative summit position to peak
start。内容和NAME_peaks.xls基本一致,适合用于导入R进行分析。
NAME_summits.bed:记录每个peak的peak summits,话句话说就是记录极值点的位置。MACS建议用该文件寻找结合位点的motif。
NAME_model.r,能通过$ Rscript NAME_model.r作图,得到是基于你提供数据的peak模型。
这一部分的工作主要是学习MACS2软件使用方法和参数的含义,难度不大。目标是找到候选靶基因。
结果注释和可视化
当我们得到上一步的输出文件后,下一步就是得到文章的结果。用的是Y叔的ChIPseeker,一个充满故事的R包,搜索一下就能看到Y叔的往事。
ChIPseeker的功能分为三类:
注释:提取peak附近最近的基因, 注释peak所在区域
比较:估计ChIP peak数据集中重叠部分的显著性;整合GEO数据集,以便于将当前结果和已知结果比较
可视化: peak的覆盖情况;TSS区域结合的peak的平均表达谱和热图;基因组注释;TSS距离;peak和基因的重叠
完全符合我们的需要,都不需要用到bedtools和deeptools。
在正式的工作开始之前,我们需要加载ChIPseeker, 基因组注释包和bed数据
# biocLite(\"ChIPseeker\") # biocLite(\"org.Mm.eg.db\") # biocLite(\"TxDb.Mmusculus.UCSC.mm10.knownGene\") library(\"ChIPseeker\")
# 根据自己存放文章具体地址修改,保证导入的数据是BED格式
ring1B <- readPeakFile(\"F:/Data/ChIP/ring1B_peaks.narrowPeak\")
cbx7 <- readPeakFile(\"F:/Data/ChIP/cbx7_peaks.narrowPeak\")
RYBP <- readPeakFile(\"F:/Data/ChIP/RYBP2_summits.bed\")
suz12 <- readPeakFile(\"f:/Data/ChIP/suz12_peaks.narrowPeak\")
得到最基本的peak数据后,我们要做以下事情
大量的韦恩图,描述不同蛋白结合基因的关系
不同蛋白在染色体结合位点与TSS的距离
为了完成上面说的两件事情,我们需要注释peak,准备TSS所在位置然后把peak比对到这些区域.
注: 原文提供不同蛋白的交集的基因TSS区域,所以要先做注释。
关于peak注释一定要多说几句:
启动子区域目前没有明确定义,一般认为基因起始转录位点的上下游区域是启动子区域,文章认为是TSS2.5 kb 以内都是靶基因
注释有两类,genomic annotation和nearest gene annotation. 前者是研究直接调控对象,后者是做基因表达调控
还有一类注释就是简单看peak上下游的范围的基因
根据文章得知需要距离TSS一定距离的注释,以及peak上下5 Kb 的注释
Genes
with a peak within the gene body, or within 2.5 kb from the TSS, were
considered to be target genes. An area of 5 kb surrounding each TSS that
was associated to one or more ChIP-seq (RYBP, Ring1B, Cbx7, Suz12, or
H2AK119ub) was used to calculate the ChIP-seq profile and the whole
ChIP-seq coverage.
ChIPseeker负责注释的就是annotatePeak,提供了3类注释方式,符合文章的要求
# 对于非向量的循环,推荐构建list,然后用lapply peaks <- list(ring1B, cbx7, RYBP, suz12) # 注释 peakAnnoList <- lapply(peaks, annotatePeak, tssRegion=c(-2500,2500), TxDb=txdb,
addFlankGeneInfo=TRUE, flankDistance=5000)
结果可以用as.GRanges或者as.data.frame查看。
as.GRanges(peakAnnoList[[1]])
as.data.frame(peakAnnoList[[1]])
得到的结果会在原先的bed文件中增加额外的注释信息,你可以和之前导入的数据进行比较,比如说geneId, transcriptId,distanceToTSS,flank_txIds等。这些都保存后续作图所需要的信息。我们可以先看不同蛋白的靶位点的注释情况,
plotDistToTSS(peakAnnoList)
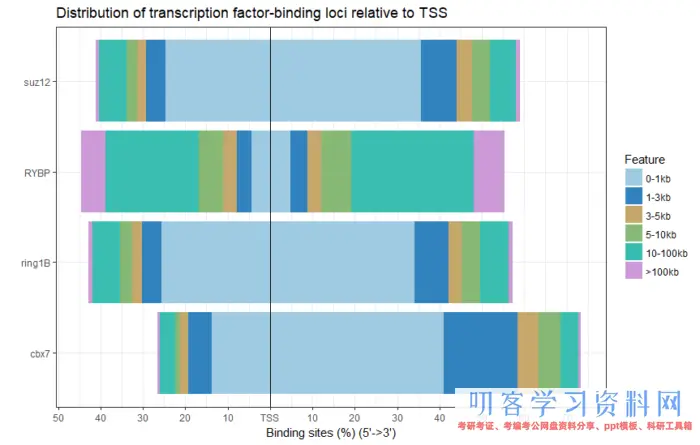
image.png
原文大量的是不同蛋白结合基因的韦恩图,我们可以用更有趣的UpSet的点图
genes= lapply(peakAnnoList, function(i) as.data.frame(i)$geneId)
library(UpSetR)
upset(fromList(genes))
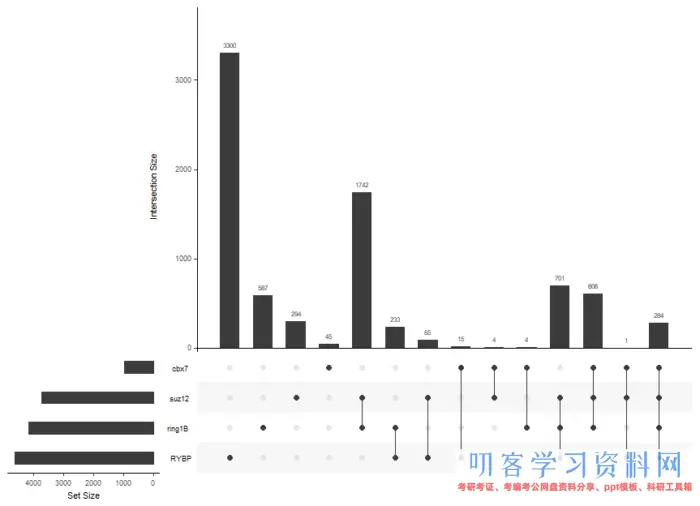
不同蛋白靶基因的集合关系
UpSet技术适用于多于5个集合表示情况,对于两个蛋白的靶基因的比较,大家更加喜欢韦恩图的。
vennplot(genes[1:2])
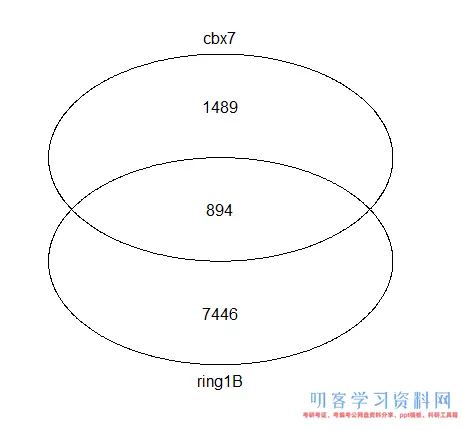
简单的韦恩图
感觉没有原文那么高大上,主要原因就是没有上色,可以自行搜索找合适的软件作图。关于不同蛋白靶基因的集合关系图基本上就是这样子。
下面是根据靶基因间的交集,绘制TSS距离图,比如说四个蛋白的重叠基因。
# 准备TSS区域,然后将peak映射到这些区域,得到tagMatrix
fourgenes <- intersect(genes[[1]],intersect(genes[[2]],intersect(genes[[3]],genes[[4]])))
target <- select(txdb, keys=fourgenes, columns=c(\"TXID\",\"GENEID\"), keytype = \"GENEID\")
promoter <- promoters(txdb, upstream = 2500, downstream = 2500)
target_promoter <- promoter[promoter$tx_id %in% target$TXID]
tagMatrixList <- lapply(peaks, getTagMatrix, window=target_promoter)
plotAvgProf(tagMatrixList , xlim = c(-3000,2999))
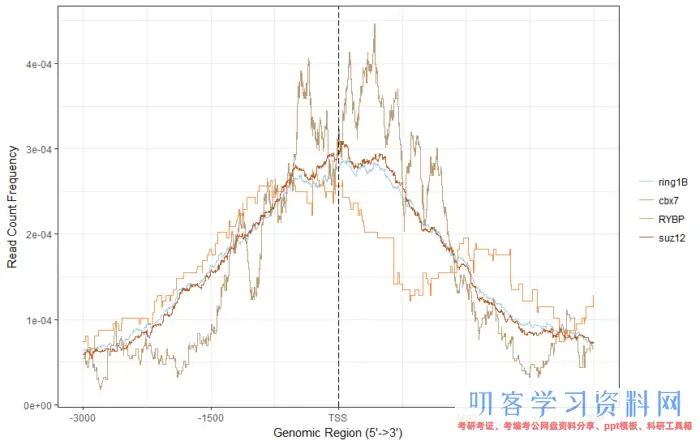
TSS距离图
感觉和原图有那么一点像,还可以画个热图
tagHeatmap(tagMatrixList, xlim=c(-2999, 3000), color=NULL)