寻找差异表达的基因并识别它们的功能,是我们进行RNA测序的最主要目的。很明显,这些差异的基因必然与功能改变密切相关,例如,比较患病个体与正常个体的组织表达谱,不难想到这些显著失调的基因参与了生物学过程、信号通路等,导致了疾病的发生。
前面已经讲了如何使用DESeq2、edgeR基于转录组测序获得的基因表达值鉴定差异表达基因。那么,后续如何继续通过生信分析的方法,探索差异表达的基因发挥了怎样的功能,参与了哪些调控通路呢?
我们平时看RNA-seq相关的文献时,文章中在鉴定了差异表达的基因后,大都会在随后承接几句关于这些失调基因所涉及通路的描述。例如,讨论这些差异基因主要映射到哪些GO或KEGG分类条目中,以说明基因表达的改变会导致哪些调控途径原有功能失调,进而与表型联系起来。通常称这种分析为GO、KEGG富集分析。
目前,能够进行GO、KEGG富集分析的工具有很多,不同的工具之间在算法、数据库组成上略有不同,因此结果也可能大相径庭。本篇就先以R包clusterProfiler的方法为例,展示如何基于给定的基因列表分析它们的GO、KEGG功能。
clusterProfiler包的安装
对于clusterProfiler的安装也很简单,一般情况下,直接通过Bioconductor安装clusterProfiler就可以了。
!!!**************************************************
#bioconductor 安装
#install.packages('BiocManager') #需要首先安装 BiocManager,如果尚未安装请先执行该步
BiocManager::install('clusterProfiler')
!!!**************************************************
clusterProfiler的GO富集分析(有参向)
首先来看GO富集分析。
注:这里均对于有参考基因组的情况而言的,无参分析暂不涉及。
1、准备输入数据
待分析的数据就是一串基因名称了,可以是ensembl id、entrze id或者symbol id等类型都可以。把基因名称以一列的形式排开,放在一个文本文件中(例如命名“gene.txt”)。Excel中查看,就是如下示例这种样式。
 输入数据,待分析的基因名称
输入数据,待分析的基因名称
2、加载参考物种的基因注释数据库
对于有参考基因组物种的分析,首先需要指定该物种的基因数据库。
存在两组情况,一种是常见物种,另一种是不常见物种。
!!!**************************************************
#(1)对于常见物种,例如人类,有些专门的 R 包数据库,例如人类参考基因组 hg19 的
library(org.Hs.eg.db)
#(2)对于不常见的物种,但却是存在参考基因组的情况
#通过 AnnotationHub 包索引基因组,例如期望找绵羊(Ovis aries)的注释库
library(AnnotationHub)
hub <- AnnotationHub()
query(hub, 'Ovis aries') #输入绵羊(Ovis aries)的名称进行匹配
sheep <- hub[['AH72269']] #返回了数据库编号 AH72269,就可以加载该库
!!!**************************************************
3、GO富集分析
加载了注释库之后,读取基因列表文件,并使用clusterProfiler的内部函数enrichGO()即可完成GO富集分析。
!!!**************************************************
library(clusterProfiler)
#读取基因列表文件中的基因名称
genes <- read.delim('gene.txt', header = TRUE, stringsAsFactors = FALSE)[[1]]
#GO富集分析
#对于加载的注释库的使用,以上述为例,就直接在 OrgDb 中指定人(org.Hs.eg.db)或绵羊(sheep)
enrich.go <- enrichGO(gene = genes, #基因列表文件中的基因名称
OrgDb = 'sheep', #指定物种的基因数据库,示例物种是绵羊(sheep)
keyType = 'ENTREZID', #指定给定的基因名称类型,例如这里以 entrze id 为例
ont = 'ALL', #可选 BP、MF、CC,也可以指定 ALL 同时计算 3 者
pAdjustMethod = 'fdr', #指定 p 值校正方法
pvalueCutoff = 0.05, #指定 p 值阈值,不显著的值将不显示在结果中
qvalueCutoff = 0.2, #指定 q 值阈值,不显著的值将不显示在结果中
readable = FALSE)
#例如上述指定 ALL 同时计算 BP、MF、CC,这里将它们作个拆分后输出
BP <- enrich.go[enrich.go$ONTOLOGY=='BP', ]
CC <- enrich.go[enrich.go$ONTOLOGY=='CC', ]
MF <- enrich.go[enrich.go$ONTOLOGY=='MF', ]
write.table(as.data.frame(BP), 'go.BP.txt', sep = '\t', row.names = FALSE, quote = FALSE)
write.table(as.data.frame(CC), 'go.CC.txt', sep = '\t', row.names = FALSE, quote = FALSE)
write.table(as.data.frame(MF), 'go.MF.txt', sep = '\t', row.names = FALSE, quote = FALSE)
!!!**************************************************
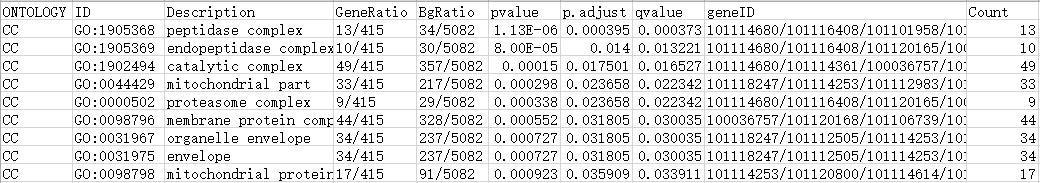 GO富集分析结果表格
GO富集分析结果表格
对于各列内容:
ONTOLOGY,GO分类BP(生物学过程)、CC(细胞组分)或MF(分子功能);
ID和Description,富集到的GO id和描述;
GeneRatio和BgRatio,分别为富集到该GO条目中的基因数目/给定基因的总数目,以及该条目中背景基因总数目/该物种所有已知的GO功能基因数目;
pvalue、p.adjust和qvalue,p值、校正后p值和q值信息;
geneID和Count,富集到该GO条目中的基因名称(分析中使用的entrze id,故这里也显示的entrze id)和数目。
注:如期望显示其它类型的基因id,如通俗的symbol id等类型,除了更改为使用symbol id的基因名称做分析外,还可以通过基因名称转换的方式对entrze id和symbol id作个匹配转换。
clusterProfiler的KEGG富集分析(有参向)
然后是KEGG富集分析。
同样地,这里均对于KEGG数据库中已经收录的物种而言的,无参分析暂不涉及。
1、准备输入数据
相比上述GO富集,clusterProfiler的KEGG富集分析方法特殊,它无需加载本地注释库,自动使用KEGG的在线数据库进行注释,因此给定的基因名称只能识别entrze
id。把entrze
id的基因名称以一列的形式排开,放在一个文本文件中(例如命名“gene.txt”)。Excel中查看,就是如下示例这种样式。
 输入数据,待分析的基因名称
输入数据,待分析的基因名称
2、KEGG富集分析
读取基因列表文件,并使用clusterProfiler的内部函数enrichKEGG()即可完成KEGG富集分析。
!!!**************************************************
library(clusterProfiler)
#读取基因列表文件中的基因名称,注意这里只能用 entrze id
genes <- read.delim('gene.txt', header = TRUE, stringsAsFactors = FALSE)[[1]]
#每次打开R计算时,它会自动连接kegg官网获得最近的物种注释信息,因此数据库一定都是最新的
kegg <- enrichKEGG(
gene = genes, #基因列表文件中的基因名称
keyType = 'kegg', #kegg 富集
organism = 'oas', #例如,oas 代表绵羊,其它物种更改这行即可
pAdjustMethod = 'fdr', #指定 p 值校正方法
pvalueCutoff = 0.05, #指定 p 值阈值,不显著的值将不显示在结果中
qvalueCutoff = 0.2, #指定 q 值阈值,不显著的值将不显示在结果中
#输出结果
write.table(kegg, 'kegg.txt', sep = '\t', quote = FALSE, row.names = FALSE)
!!!**************************************************
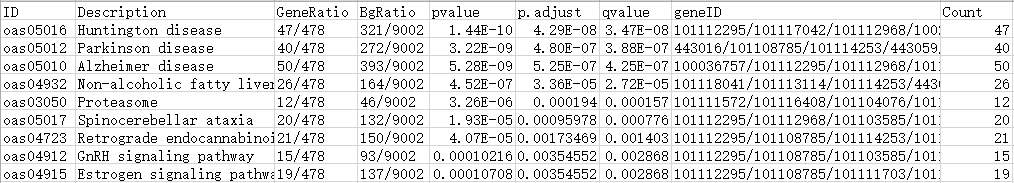 KEGG富集分析结果表格
KEGG富集分析结果表格
对于各列内容:
ID和Description,富集到的KEGG id和描述;
GeneRatio和BgRatio,分别为富集到该KEGG条目中的基因数目/给定基因的总数目,以及该条目中背景基因总数目/该物种所有已知的KEGG功能基因数目;
pvalue、p.adjust和qvalue,p值、校正后p值和q值信息;
geneID和Count,富集到该KEGG条目中的基因名称(分析中使用的entrze id,故这里也显示的entrze id)和数目。
注:如期望显示其它类型的基因id,如通俗的symbol id等类型,由于该分析中只能输入entrze id,因此可以通过基因名称转换的方式对entrze id和symbol id作个匹配转换。
clusterProfiler附带的作图功能
此外,clusterProfiler中也额外提供了一系列的可视化方案用于展示本次富集分析结果,具有极大的便利。
!!!**************************************************
#clusterProfiler 包里的一些默认作图方法,例如
barplot(kegg) #富集柱形图
dotplot(kegg) #富集气泡图
cnetplot(kegg) #网络图展示富集功能和基因的包含关系
emapplot(kegg) #网络图展示各富集功能之间共有基因关系
heatplot(kegg) #热图展示富集功能和基因的包含关系
!!!**************************************************
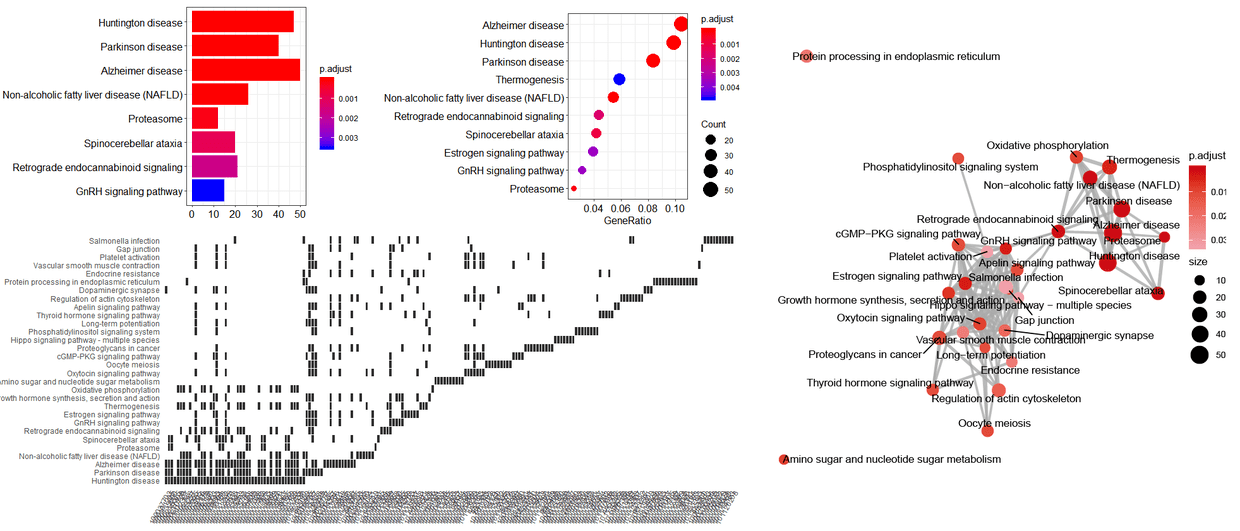 一些富集柱形图、气泡图、网络图等
一些富集柱形图、气泡图、网络图等
!
!
!
注:!!!*******之间为R脚本内容。
搜索微信公众号“纪伟讲测序”